In the fourth quarter of 2009, there were more than 800 agents in clinical trials for the treatment of cancer,1 more than the number in any other therapeutic drug class.2 The hematology/oncology therapeutic category comprises more than one third of all drug development candidates by some estimates.2 Compared to many areas of medicine, the opportunity to identify additional novel molecular targets for cancer appears especially promising. To date, 350 cancer genes have been identified. It is possible, however, that researchers applying advanced DNA-sequencing techniques will identify more than 2,000 cancer genes, each offering a potential for multiple new agents.3
The Challenge of Cancer Drug Development
The sponsor of each new investigational agent ultimately seeks approval by major regulatory agencies, including the U.S. Food and Drug Administration (FDA), to market their product for at least one indication. However, only 2 to 7 new drugs and biologics for the treatment of cancer attain this goal per year. Marketing approval represents the final hurdle that sponsors must clear in their time-consuming efforts to discover and optimize lead compounds, test new drugs in animals, and conduct clinical studies in man. Without this license, the effort to develop a new drug, which may have consumed hundreds of millions of dollars and spanned more than a decade, could go in vain.4,5 Small companies that concentrate on cancer drug development and which fail to attain marketing approval for at least one of their agents will eventually lose access to capital and will be forced to wind down operations or solicit an offer to be acquired. The impact of failures at larger companies is typically less extreme, but there is still a steep opportunity cost of failure that sponsors seek to avoid.
Companies face low odds, high expense, and long planning cycles in their quest to develop new agents. Fewer than one in ten investigational agents that enter phase 1 trials ultimately gain marketing approval.6 The low odds of success is a major contributor to the expense and risk associated with cancer drug development.7,8 The most recent published estimate of the average total cost (i.e., preclinical plus clinical costs) of developing a representative new cancer drug from concept to FDA approval is $1,042 million (in $2,000).8,9 This figure accounts for the cost of developing other drugs that fail as well as the opportunity cost of capital. The average cost to develop a new oncology drug exceeds the mean of other therapeutic classes by approximately 20% and is almost twice the cost of a drug for HIV/AIDS.10 The higher costs to develop cancer drugs compared with other therapeutic classes are due in part to a greater frequency of late-stage failures and longer development times.5,10 Sponsors can generally develop highly active cancer products quickly, but many compounds are not highly active and require time and careful planning to demonstrate their potential. It takes 14 years, on average, for a company to take a new anticancer agent along the path from preclinical testing through phase 3 human trials, with approximately half of the time spent in clinical development.5,6,8
Developing cancer drugs has always proven to be a challenging undertaking. The combination of an incomplete understanding of cancer biology, poorly predictive animal models, and frequently uninformative phase 2 trials has limited the speed of advancement over much of the last 50 years. The era of molecularly targeted therapies has brought not only enormous optimism and opportunities for advancement but also additional challenges. There is now an appreciation of genetic heterogeneity among most populations of cancer patients, yet investigators have encountered scientific and logistical impediments in their attempts to use biomarkers to enrich trials for patient subsets most likely to respond to the new agents. There has also been lack of consistent agreement on the definition of clinical benefit, which in turn has created uncertainty for sponsors and required ongoing collaborative input from multiple stake-holders. In some instances, the understanding of the transformation process and cancer phenotype is now greater than the ability of clinical researchers to evaluate efficiently the clinical potential of new drugs.11 Some researchers have even suggested that drug development principles that evolved with traditional cytotoxic agents may not apply in the era of targeted therapy, since the newer agents possess different mechanisms of action and antineoplastic effects and have a different spectrum of toxicity.12 It seems more appropriate, however, to build on the established principles and development platform, modifying them where necessary to account for methods of patient enrichment and adding new tools to assess biological and clinical activities of the new agents.
The Commercial Opportunity
Despite the risk and complexity, cancer drug development efforts have attracted substantial amounts of public and private funding, driven by the continued unmet medical need of cancer patients and potential for blockbuster success. In 2008, the worldwide market for cancer drugs was approximately $68 billion.13 Over the 20-year period from 1987 to 2008, worldwide sales of cancer drugs grew at an annualized rate of 11%, exceeding by a wide margin the growth in sales of other major therapeutic categories in medicine. The growth in cancer therapies continues to be among the highest in the major therapeutic classes. The widespread adoption of the newer targeted agents has driven much of the market growth. Anticancer products like imatinib mesylate (Gleevec), bevacizumab (Avastin), and rituximab (Rituxan) now rank among the top-selling prescription agents across all drug categories, with 2008 worldwide sales of $3.7 billion, $5.0 billion, and $5.6 billion, respectively.13 Research analysts predict that the size of the oncology market will grow to approximately $90 billion by 2014. This growth will likely make the oncology sector the largest therapeutic market within 5 years, overtaking sales of cardiovascular medicines. Every large pharmaceutical company and countless smaller biotech companies have major efforts in cancer drug development. Analysts estimate that monoclonal antibodies and oral tyrosine kinase inhibitors alone will reach $22 billion in worldwide sales by 2012.
Spending on cancer drug development has also grown more quickly than overall spending on biomedical research, which itself has more than doubled in real terms over the last 10 years.6 Some analysts predict that companies will advance more than 500 anti-cancer agents into clinical development within the next 10 years,14 consuming billions of dollars in public and private research capital in addition to patient resources. Pursuing efficient clinical development strategies is therefore not only an important issue for sponsoring companies but also a matter of public interest. This chapter will review various stages and strategies for the development of cancer drugs as well as regulatory hurdles for their marketing approval.
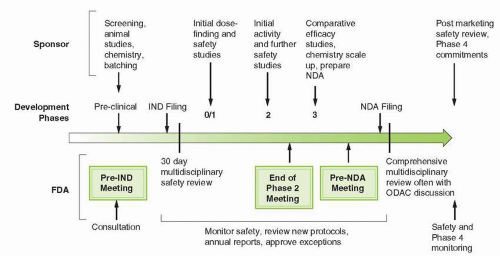
FIGURE 3-1 Overview of the drug development process. The multiple interactions between drug sponsors and the FDA reflect the convergence of the duties and responsibilities throughout the phases of development. IND, initial new drug application; NDA, new drug application; ODAC, Oncologic Drug Advisory Committee.
The Process of Cancer Drug Development
Clinical investigators and sponsors collaborate to develop cancer drugs in a stepwise process of discrete clinical phases, each with their own endpoints and milestones (Fig. 3-1). Cancer researchers have built the development process over many years to establish safe dosing, assess efficacy, and determine relative effectiveness compared to reference regimens. The process continues to evolve as investigators recognize the need to adapt clinical development paths to the more targeted nature of the newer agents. Over the past 5 years, clinical trial designs have become more flexible and innovative and have begun to utilize tissue genotyping and other biomarkers to select for patients who are most likely to receive a clinical benefit. These changes reflect the types of drugs under investigation. Until recently, almost all anticancer drugs were cytotoxic or hormonal. Since 2005, more than 60% of cancer drugs approved by the FDA have been molecularly targeted therapies.15 The targeted therapies have thus far fallen into three major categories. Of the 169 targeted agents either on the market or in late-stage clinical development, 84 (50%) are small molecule kinase inhibitors, 30 (18%) are small molecule inhibitors of other types of enzymes implicated in cell growth, and 55 (32%) are large molecule biologics, including monoclonal antibodies.15 There is reason to believe that sponsors will continue to adapt the process of cancer drug development to the types of agents under investigation. The biotech industry, comprising smaller, more focused firms, now accounts for the majority (68%) of cancer drugs in clinical development. These firms tend to display more flexibility in their development strategies and may face more scrutiny and pressure by investors to develop the agents as quickly and as efficiently as possible.
Sponsors decide whether to advance an investigational agent into clinical testing on the basis of overall interest in the agent’s target, the strength of preclinical data in cell lines and animal models, and the attractiveness of the agent’s pharmaceutical properties. Most companies will also perform some analysis to determine whether the expected return on investment exceeds a predetermined hurdle-rate, which is often similar to the firm’s estimated cost of capital. A broad discussion of the preclinical problem of target validation and assessment is beyond the scope of this chapter (see Chapter 2). However, if the target is not essential to growth, spread, or survival of the tumor, or if pathway redundancy exists that allows the tumor to escape from single pathway inhibition, the potential therapy is not likely to produce a clinical benefit.5,16
Once a sponsor has decided to pursue clinical development, a number of regulatory and scientific review steps must be completed prior to initiating clinical trials. These requirements can prove onerous for many sponsors, but they help to protect research subjects participating in trials, focus the research effort, and, if successfully employed, may improve data quality.17 In their planning, sponsors collaborate with academic investigators, the FDA, and in some cases, the National Cancer Institute (NCI) to decide on the optimal patient populations to study, appropriate trial endpoints and designs, and potential for combination regimens with other approved or investigational agents.18 The number of permutations of possible developmental strategies available to a sponsor for any given investigational agent is enormous, but a thorough understanding of the underlying biology can help clarify the overall development path and avoid unnecessary detours and delays.
Phase 0
Clinical investigators, FDA, and the NCI have all recognized that a major weakness inherent in the cancer drug development process is the preclinical/clinical transition.5 Most agents fail in clinical testing because animal studies fail to predict their behavior in humans. Tumor-bearing mice have been useful in determining antitumor activity of target inhibitors, comparing drug analogues, and optimizing dose and schedule of administration. Unfortunately, however, the mouse does not predict reliably for success in the clinic. Drug disposition is in general faster, tolerance for toxicity greater, and growth of tumors more rapid in the mouse, all of which tend to make drugs more effective in mice. Although extensively employed in preclinical drug testing, neither human tumors grown in immunodeficient mice nor spontaneous murine solid tumors have reliable predictive value for human tumors.19 Often single animal or xenograft tumors are subjected to testing, but the results have unclear relevance to the diversity of molecule subtypes encompassed by a single pathological category (colon cancer) in humans.
Up to 25% of investigational cancer agents that enter phase 1 trials will be discontinued prior to phase 2 studies.6 About half of these early discontinuations are due to undesirable pharmacokinetic (PK) characteristics that are inconsistent with the predictions made from preclinical animal models.11 It would therefore be helpful if investigators had access to additional PK and pharmacodynamic (PD) data early in the development effort to help ensure that the investigational agents are interacting with the desired targets and in the appropriate concentrations prior to initiating more expensive and time-consuming phase 1 and 2 trials. When sponsors do not know until well into the phase 2 effort whether an investigational agent displays the desired biological activity or interacts with the intended target, resources may be spent on programs with little promise, displacing the opportunity for more worthwhile investments.
In 2004, the FDA undertook an effort to modernize the clinical trials process called the Critical Path Initiative. As one of the first changes under the Critical Path, the FDA introduced new guidelines for early exploratory drug studies in order to improve the preclinical/clinical transition. A 2006 guidance document20 provides specific recommendations regarding safety testing, manufacturing, and clinical approaches to be used in very early clinical studies, sometimes called exploratory or phase 0 trials. The goal of phase 0 studies is to establish quickly whether an investigational agent displays biological activity in humans before large numbers of patients receive exposure to potentially inactive agents.14 The phase 0 strategy requires fewer preclinical animal studies than a typical phase 1 trial and allows sponsors to make smaller batches of an experimental drug.21 To mitigate safety issues that may arise in exploratory testing, researchers limit these early human tests to a few patients (10 or fewer), shorten the time of drug exposure (e.g., 7 days or less), and reduce the doses of medicines than would be employed in a typical phase 1 trial (e.g., <1% of what researchers expect would be the agent’s standard dose).14 Types of phase 0 trials include microdose studies evaluating PK or functional imaging modalities as well as trials evaluating mechanisms of action related to efficacy. Phase 0 studies may evaluate and compare a series of related compounds.11 Functional imaging studies or target measures in normal tissues may be useful when repeat biopsies before and after dosing are not possible or logistically difficult. Importantly, phase 0 trials are neither intended to replace the traditional dose escalation, safety, and tolerance studies that are required in phase 1 testing nor intended to evaluate clinical benefit.
It would be inappropriate for researchers to employ a phase 0 trial for each new agent. First, a small dose of an investigational agent may not provide meaningful information in the absence of a validated biomarker. At present, most new investigational agents do not have a validated biomarker that can predict tumor activity with sufficient accuracy at the start of clinical development to make optimal use of a phase 0 trial.21 Second, researchers must be sure that the drug’s mechanism of action is defined by that target to avoid misleading data.14 Sorafenib, for example, was originally tested in clinical trials as a B-Raf kinase inhibitor, but its primary mechanism of action in renal cell carcinoma may be the result of other “off-target” effects on angiogenesis. Third, the PK inferences drawn from very small doses of drugs are reliable only if the drug displays linear or first-order pharmacokinetics (the plasma concentration of drug at a given time after dosing is directly proportional to the administered dose). The majority of clinically used cancer agents display linear PK; however, those that are eliminated by a potentially saturable process such as active tubular secretion or enzymatic metabolism are unlikely to be good candidates for phase 0 testing. Therefore, the ideal agent for phase 0 testing has a target that can be monitored, a biomarker that can be assayed with validity and reproducibility, and kinetics that appear linear.
The recent trial involving ABT-888, an orally bioavailable inhibitor of poly (ADP-ribose) polymerase (PARP), provides an excellent example of a phase 0 trial.22 PARP inhibitors may hold particular promise as chemotherapy and radiation sensitizers since PARP is involved in DNA repair via poly (ADP-ribosyl)ation of histones and DNA repair enzymes. The primary endpoint of the trial was PD, specifically target modulation in human samples. The effort was successful: within 5 months of starting the phase 0 trial, strongly supportive data were available including the molecular proof-of-target inhibition by ABT-888, as well as PK and PD data that could serve as the foundation for subsequent combination studies of ABT-888 with DNA-damaging agents. Specifically, the data demonstrated that a twice-daily schedule for ABT-888 was appropriate, and, based on significant inhibition of PARP in tumor biopsies at 25 mg, the authors recommended that the phase 1 dose of ABT-888 in combination with DNA-damaging agents should be 10 mg BID. ABT-888 was an appropriate candidate for testing in a phase 0 trial because its preclinical data demonstrated that PARP inhibition was indeed the target for ABT-888, and there was a validated assay before the initiation of phase 1 trials. Additionally, researchers demonstrated that PARP inhibition, needed for preclinical antitumor activity, occurred at doses and exposures well below those associated with toxicity.11
This example highlights several important points about phase 0 trials. First, the rapid completion of complex, early-phase clinical trials requires an integrated, multidisciplinary research team capable of performing PK and PD studies in real time. The authors of the ABT-888 accomplished their studies in 48 hours or less after sample acquisition. Second, researchers must pay particular attention to ensure that biopsy procedures are performed only after careful validation of the assay procedures. The authors of the ABT-888 study made efforts to ensure that that they could draw statistically and scientifically meaningful inferences from a limited number of biopsy samples. Finally, researchers should make efforts to avoid unnecessarily invasive studies if possible. The authors of the ABT-888 study found that there was a strong correlation between the effects of ABT-888 in peripheral blood mononuclear cells (PBMCs) versus tumor samples, which may obviate the need for biopsies in future clinical research. It is possible that examples such as this one will drive the adoption and broader uptake of carefully conceived, PD-driven driven early-phase clinical trials in oncology.22
Several sponsors such as Novartis, Merck, and Pfizer have registered phase 0 clinical trials in the ClinicalTrials.gov database. They are using these trials to explore PK and PD primary objectives as well as positioning backup compounds. At the very least, phase 0 trials may help clinical investigators choose among several potentially promising compounds before sponsors are required to undertake the requisite small and large animal preclinical work.11 Whether or not sponsors choose to incorporate phase 0 trials, researchers should generate sufficient preclinical data prior to phase 1 in order to (a) establish the molecular target of the novel drug; (b) determine the effect of the drug on normal and malignant cells; and (c) explore the relationship between the agent’s dose/schedule and its antitumor effects, target effects, PK measures, and toxicology.12 This work will help to define the optimal biological dose in phase 1 and may allow investigators to incorporate analytically validated assays into early phase trials of the agent to assess for target modulation.18
Phase 1
There are more than 1,000 phase 1 intervention studies open world-wide according to ClinicalTrials.gov, and the numbers are steadily increasing as more products from the biopharmaceutical industry reach the clinic.23 Traditionally, phase 1 trials have represented the first testing of investigational agents in humans. The two major objectives of these trials are to characterize the agent’s toxicity profile and determine a dose and schedule appropriate for phase 2 testing. Under the standard phase 1 design, investigators enroll successive cohorts of 3 to 6 patients at increasing doses of an experimental therapy with the goal of determining dose-limiting toxicity and then backing down to a dose appropriate for phase 2 testing.24 Phase 1 trials in most other areas of medicine enroll healthy participants, whereas phase 1 trials in oncology typically enroll patients who have advanced cancer and who have exhausted standard treatment options. A second difference among oncology-focused phase 1 trials is that sponsors and physicians also seek to evaluate the potential for therapeutic benefit, usually as a secondary endpoint.24, 25, 26 Such observance for therapeutic benefit in phase 1 is appropriate from a development standpoint, given the positive correlation found between response rates (RRs) in phase 1 and ultimate FDA approval.24
The more recent focus on modern molecularly targeted agents has drawn into question several aspects of traditional phase 1 trials. Toxicity has been the classical primary endpoint used to determine the recommended dose for phase 2. The basis for recommending the maximum tolerated dose (MTD) for phase 2 testing focuses on the assumption that the therapeutic effect and the associated toxic effects are correlated and that the mechanism of action of the therapeutic and that of the associated toxic effects are correlated. Such an assumption may not be appropriate for the new generation of targeted agents.18 These agents often have a wider therapeutic index; the proposed mechanism of therapeutic versus toxic effects could differ due to off-target effects; they may require prolonged doses at relatively low levels to provide clinical benefit; and they may not cause impressive tumor shrinkage as single agents.18 Some investigators have even expressed a view that the key point in phase 1 trials for targeted therapies should evolve from the current idea of MTD in normal tissue to a more suitable endpoint of the dose required to maximally inhibit the relevant target in tumor tissue.27 Moreover, most sponsors now realize that demonstrating that a novel compound has the target effect for which it was designed may be an equally important aspect of phase 1, particularly if such target validation was not achieved prior to a phase 1 effort.12
Use of Biomarkers in Phase 1
One of the more controversial trends in phase 1 trials is the inclusion of biomarker assays. Goulart et al. found that of 2,458 abstracts of phase 1 clinical trials submitted to the American Society of Clinical Oncology (ASCO) annual meetings from 1991 to 2002, approximately 20% (503 of 2,458) included at least one biomarker study as part of the phase 1 trial. This proportion increased over time (14% of the abstracts reported inclusion of biomarkers in 1991 compared with 26% of the abstracts in 2002).28 Trials focusing on large-molecule biologics and those sponsored by the NCI were more likely to include a biomarker study as part of the trial. Biomarkers contributed to dose and schedule selection in only 13% and 8%, respectively, and in only one instance out of 87 trials did a biomarker contribute to dose selection independent of clinical (i.e., MTD) data. However, biomarkers played a more substantive role in confirming mechanism of action (39% of the trials) and in helping to select patients for future studies (22%). Biopsies of either normal or tumor tissue were more likely to confirm mechanism of action compared to serum-or imaging-based biomarkers. Similar findings were seen in a review of phase 1 trial designs for solid tumor studies that focused exclusively on targeted, noncytotoxic agents.29 Of the 60 studies reviewed, 36 used toxicity data and 8 used PK data as endpoints for selection of the recommended phase 2 dose. Biomarker studies provided the basis for dose selection in only one trial that evaluated an epidermal growth factor receptor (EGFR) antibody. Drug effects on surrogate tissue (e.g., PBMC or buccal mucosa) provided the primary basis for the recommended phase 2 dose in only one study, which evaluated a Raf kinase inhibitor. In ten (17%) of the trials, however, biomarker studies in surrogate tissue did provide supplementary data supporting dose selection. Information from functional imaging studies was included in six trials, and it provided the basis for a recommended phase 2 dose in one trial.
The phase 1 trial of the PARP inhibitor AG014699 in combination with temozolomide provides an example of the potential promise and limitations of including biomarker studies in phase 1 trials.30 Preclinical evidence demonstrated that inhibiting PARP potentiates the effects of cytotoxics, particularly alkylating agents and topoisomerase 1 inhibitors. Investigators were able to establish, within the context of the phase 1 trial, the dose of AG014699 that effectively inhibited PARP in peripheral blood leukocytes and confirmed the inhibition in tumor deposits from melanoma. They then used the PD endpoint of PARP inhibition, rather than the more classic toxicity or PK variables, to define the dose recommended for phase 2 trials. Although successful, this trial also demonstrated the potential limitations of using blood-based tissue to establish a PD-defined dose. For example, the investigators found no evidence of increased PARP inhibition between the dose levels of 12 and 18 mg/m2 AG014699 in the surrogate PD tissue (PBMC), whereas they did observe a trend toward a dose-dependent increase in inhibition in tumor biopsies. Second, many targets for cancer such as PARP are overexpressed in malignant tissues, so it is possible that doses sufficient to inhibit PARP in surrogate tissue may not be high enough to do so effectively in tumor tissue. Another recent example of a biomarker that was studied in phase 1 is the translocation of the EML4-ALK gene, which appears to predict treatment response to a kinase inhibitor targeting ALK. A phase 1 study reported that ten out of 19 (53%) patients with the translocation had a significant response to treatment with a targeted oral drug that inhibits ALK receptor kinase (PF-02341066). This very promising finding, which defines a new subset of patients with non-small-cell lung cancer (NSCLC), awaits confirmation in more advanced trials.31 The ALK receptor kinase trial demonstrates that when investigators apply effective biomarkers in phase 1, the inferences gained from these early trials may be equal to or exceed what can be produced from traditional phase 2 studies.
In 2007, investigators from the NCI and the NCI of Canada Clinical Trials Group led a task force along with other academic researchers to review design issues in phase 1 studies of targeted anticancer agents.12 The group specifically addressed whether toxicity (i.e., MTD) and PK data should remain the focus of phase 1 testing. They concluded that both toxicity and PK remain appropriate endpoints to establish the dosing range for novel compounds, advising that investigators establish dose ranges with the upper limit defined by toxic effects, particularly if toxic effects are mechanism based. If no toxic effects are seen, the group recommended other factors to consider including biomarker measurements, PK measures, and feasibility of delivery of the dose. The task force concluded that the recommended dose should be the highest safe dose unless there is a clear biological rational otherwise.
The group also addressed the issue of whether investigators should routinely include biomarker studies in phase 1 trials, and in general they raised a number of cautions.12 In particular, they recommended that any planned biomarker assays should be developed and fully validated before the start of the phase 1 study and that such studies should preferentially be focused in patients enrolled at potentially therapeutic doses so that they may be helpful in establishing a “biological active” dose range. Finally, the group cautioned that potentially hazardous/uncomfortable procedures should not be mandated unless essential to the endpoint of the study, despite evidence that tumor biopsies can be performed safely in most instances in the context of phase 1 trials.27 They suggested that when molecular proof of principle appears important for subsequent development decisions, clinical investigators should consider expanding one or more cohorts after the conclusion of the escalation phase, or designing a separate study to confirm that the doses identified on the basis of toxicity in fact are able to affect the molecular target.12
It would be ideal to have obtained evidence of antitumor effect and to have identified a biomarker at the completion of phase 1 to help inform phase 2 trials. However, whether or not biomarker tests are employed, the phase 1 effort should have determined a phase 2 dose recommendation, a range of doses that appear biologically active if future combination studies are planned, and a decision whether to proceed with further development based on prespecified criteria that may include evidence of intended biological activity).29Table 3-1 presents a proposed generalized prephase 3 clinical development plan for antineoplastic agents.
TABLE 3.1Generalized prephase 3 clinical development plan for antineoplastic agents
Phase 1
Explore does range up to MTD unless limited by formulation or bioavailability
Include as heterogeneous a patient population as can be ethically justified
Define relationship of dose to toxicity and PKs
Observe for mechanism-related toxicity (e.g., skin rash with EGFR inhibitors) as a readily observable biomarker
Consider inclusion of biomarker studies on readily accessible tissue (e.g., peripheral mononuclear cells) to assess mechanism of action and minimal potential effective dose
Consider use of tumor biopsies at highest dose if results are to be used to make “Go/No Go” decision
Phase 2a
Unless anticipated evidence of benefit is partial response, consider conducting dose-ranging randomized trials to detect activity, including use of novel endpoints
Consider crossover design to allow for use of placebo when possible
Consider analysis of previously collected diagnostic material to correlate with activity
Phase 2b
Consider studies of predictive biomarkers (including tumor biopsies) once drug is shown to be active
Adapted from Ratain MJ, Glassman RH. Biomarkers in phase I oncology trials: signal, noise, or expensive distraction? Clin Cancer Res 2007; 13(22 Pt 1):6545-6548.
Phase 2
There are more than 1,200 phase 2 intervention studies open worldwide,23 evaluating almost 500 new agents.32 The primary focus of phase 2 trials is to determine efficacy in different tumor histologies or molecular subtypes of cancer.33 When successful, phase 2 trials usually present the first opportunity for investigators to demonstrate evidence of efficacy in humans. Compared to phase 1 trials, these studies are larger (30 to 70 patients), utilize potentially therapeutic doses and schedules, and enroll patients who are less heavily pretreated. The classic phase 2 oncology trial is a single-arm, open-label intervention study. Investigators compare the trial results to historical controls in order to make a “go/no go” decision whether to progress onto definitive phase 3 studies. In order to effectively design phase 2 studies, investigators must take into consideration the expectations for biological activity of the target agent (tumor shrinkage versus stabilization), the level of target modulation of the target documented in previous studies (phase 0 and 1 studies), appropriate patient selection, and early determination of activity (e.g., functional imaging) for a given target.18 The probability of an investigational agent progressing to a phase 3 once it has entered phase 2 trial is approximately 40%.34
From a statistical perspective, most phase 2 trials are screening studies that attempt to identify agents most appropriate for phase 3. Most sponsors design phase 2 trials to maximize the chance of detecting clinical response or biological activity and make every attempt to avoid erroneously concluding that the agent is inactive (i.e., type II error).33 Typically, investigators have focused on whether an investigational agent has a minimum activity level in patients with good performance status and minimal prior chemotherapy, rather than trying to design optimal trials to predict for phase 3 success.35 Tumor response, measured by the response evaluation criteria in solid tumors (RESIST)36 and expressed as a proportion of the total number of patients treated, has typically been the primary outcome. Researchers then compare the trial outcome to a prespecified target,37 determining the target by specifying a “null” and an “alternative” proportion of responses. In the widely used Simon optimal design,38 a null RR of 5% and an alternative of 25% require a target of at least four responses in 30 patients.37 Under another design, the trial is suspended if there are no responses in the first 12 evaluable patients, since there would be less than a 10% chance of rejecting a drug that has a true RR of 20%.33 Sponsors chose the null and alternative RRs based on multiple considerations, including historical RRs and the availability of alternative therapies in the particular setting under investigation.
Clinical researchers have debated the overall utility of phase 2 oncology trials. Some have argued that phase 2 oncology trials are not as predictive of phase 3 success as in other areas of medicine, in large part due to their lack of randomization.39 Others have even suggested that progression from phase 1 directly to phase 3 may be appropriate for targeted agents, given that novel agents can show a survival improvement in phase 3 with little or no evidence of response in phase 2 (e.g., sorafenib in kidney cancer). In one review of 43 phase 3 studies published from 1998 to 2003, the authors were unable to identify any phase 2 study characteristics that predicted significantly for a “positive” phase 3 study, though there was a trend toward larger sample sizes in phase 2 (and therefore more accurate prediction of response) being associated with a greater chance for a “positive” phase 3 study.40
Other data support the utility of phase 2 trials in the evaluation of both cytotoxic and targeted agents. In a review of 46 cytotoxic investigational agents studied in 499 phase 2 trials from 1985 to 1999, higher overall RRs (particularly above 20%) in phase 2 trials did predict for regulatory approval in most types of solid tumors (with the exception of melanoma and renal cell carcinoma).41 This relationship between response in phase 2 and regulatory approval has also held for some targeted agents. In a review of 89 single-agent phase 2 trials involving 19 targeted drugs in six solid tumors obtained from a database of targeted agents whose phase 1 trial was published in 2004, objective responses were seen in 38 trials. In 19 trials, RRs exceeded 10%, and in eight trials, they exceeded 20%. Similar to what was seen among cytotoxic agents, higher overall RRs were predictive of regulatory approval in the tumor types reviewed.42 The authors conclude that even with targeted agents, a lack of any evidence of response in phase 2 suggests that the drug is likely to fail in subsequent development. Despite the conflicting data on their utility, phase 2 screening trials remain critical to the process of choosing which agents to advance to definitive testing. Without their application, several thousand large-scale (phase 3) trials would be required to evaluate all the potential new agents and their combinations, which is clearly not feasible from an ethical or economic standpoint with the currently available resources and with the available pool of patients who are willing to enroll in clinical trials.5,43 Investigators have recently focused on two strategies to improve the utility of phase 2 studies: patient randomization and enrichment.44
Randomization in Phase 2
Investigators have not, until recently, included patient randomization in phase 2 trials, choosing to employ historical comparisons instead. In one review of all phase 2 trials published in 2002, including 586 cancer studies, only 7 (<1%) of the trials conducted by oncology specialists were performed with a randomly selected control population that was blinded to the intervention.32 Investigators have also not consistently applied a systematic approach to analyzing historical data. In a review of 251 phase 2 trials published from 2003 to 2005, nearly half (46%) of the papers failed to cite the source of historical data used for comparison, and no study used statistical methods to account for either sampling error or differences in case mix between the historical cohort and the phase 2 sample.37 Uncontrolled phase 2 trials of combination therapies have been even more difficult to interpret, as it is impossible to quantify the additional response to the underlying therapy and to estimate the potential incremental improvement to longer-term outcomes such as overall survival.43 For these reasons, investigators have chosen to include randomization in more phase 2 trials. Some researchers have even suggested that phase 2 randomization be mandatory in the evaluation of molecular targeted agents, especially for the development of drug combinations.39
The use of randomization in phase 2 can be justified under certain conditions, but its broad application has limitations. The major justification for randomization is that valid historical benchmarks may not exist for certain endpoints such as progression-free survival (PFS) or for certain subsets of diseases, especially for groups of patients expressing a particular marker or target. In these instances, investigators may randomly assign treatments to subjects in order to minimize bias and therefore gain confidence in the inferences drawn from the trial data. There are, however, a number of methodological concerns regarding the interpretation of relatively small, randomized phase 2 trials.44 First, if endpoints are not carefully planned and considered, randomized phase 2 trials may be rendered as underpowered phase 3 trials. Second, randomized designs may require up to four times as many patients as single-arm studies, which are then compared to historical controls, but with similar statistical operating characteristics.35 For this reason alone, randomization will be less efficient when an adequate historical database is present. There is also more expense involved (and more time spent) in the assessments necessary to evaluate and confirm tumor progression when a PFS endpoint is chosen, and investigators may have difficulty enrolling randomized studies compared to single-arm trials (particularly if a placebo is used in the control arm). Given these limitations, a recent task force of academic investigators concluded that single-arm phase 2 studies with traditional statistical designs remain appropriate, even with newer targeted agents, as long as the investigational agents are tested in tumor types for which a robust historical database exists (e.g., NSCLC) or in salvage settings where no standard treatment exists (e.g., cholangiocarcinoma).44
Phase 2 Enrichment Strategies
Enrichment strategies represent the second major modification that investigators have made to phase 2 trials in recent years. The underlying goal of an enrichment strategy is to increase the likelihood of detecting an effect of a drug by limiting eligibility to patients who are most likely to respond or by excluding those unlikely to respond based on clinical characteristics or biological features of the tumor.16 The motivation for enrichment stems from genomic and proteomic studies that have demonstrated that most cancers are heterogeneous. Clinical investigators have in turn focused on using molecular (e.g., tumor genotype of level of gene expression) or imaging biomarkers to stratify patients beyond tumor histology in order to direct targeted agents to those patient subsets most likely to receive a benefit.45 Enrichment designs may be particularly useful when the prevalence of the target is low compared to the overall populations, the target assay is very accurate, and the therapy is expected to have little effect in nontargeted patients (e.g., trastuzumab in breast cancer). When one of these elements is missing or uncertain, attempts at enrichment may be misleading and it may be more appropriate to consider a design that evaluates treatment effects on both the overall population and specific patient subsets. Sponsors must cooperate with investigators to balance the enrichment strategy’s potential to increase the success rate of new drug development against the added cost and regulatory complexity of attempting to simultaneously develop a drug and companion biomarker test.16
There are two major types of enrichment designs: those that require a putative validated biomarker and those that seek to enrich for benefit using trial designs. The term biomarker in the context of phase 1 studies has generally been used to describe a measure of PD effect (e.g., dynamic contrast enhanced-magnetic resonance imaging [MRI] suggesting angiogenesis inhibition). For phase 2 and 3 studies, the term is used differently, referring instead to markers (either tissue based or serial measures of surrogate effects such as changes in functional imaging) that predict for a treatment effect (either benefit or lack of benefit).44 The striking and rapid development path for imatinib in patients with the novel fusion gene, BCR-ABL, and the decision to limit the registration trial of trastuzumab to those patients with breast cancer who overexpress the HER2/neu receptor provide excellent examples of biomarker-based enrichment strategies.46,47
Such biomarker-based enrichment approaches are preferred if they are available, but several factors limit their broad application. First, investigators need to have a full understanding of the drug target and the assay used in its evaluation. Unfortunately, such an understanding is often not achieved until after the agent completes phase 3 testing and marketing registration.16 Early clinical trials of cetuximab for the treatment of colorectal cancer, for example, mandated that the patient’s tumors express the EGFR in an attempt to enrich for patients most likely to receive a benefit. However, the trials showed little relationship between EGFR expression and the probability of tumor response.48 Second, if the drug has pleiotropic effects or the marker does not correlate well with the biological activity of the relevant pathway, the enrichment strategy is likely to fail.16 If investigators had focused exclusively on sorafenib’s original putative target, Raf kinase, they may have been distracted from its ultimate utility in advanced renal cell cancer, which likely relates to its effect on vascular endothelial growth factor (VEGF).49 Third, there may be molecular differences between primary tumors and metastatic disease. For example, the estrogen and progesterone receptor and HER-2/neu status are usually known for each patient with breast cancer. The status, however, is typically based on analyses of the primary cancers, even when metastatic disease is treated. The level of discordance, gain or loss, of these markers has been shown to be as high as 20% or more for HER-2/neu.50 Finally, in many instances, the scientific progress required for efficient biomarker-based development has lagged. For example, neither vascular VEGF nor EGRF expression correlates well with response to their respective antagonist.49 Given these current limitations, biomarker-based approaches will require optimization before they can have a broader impact on cancer drug development.
Only gold members can continue reading. Log In or Register to continue