Prognostic and predictive factors and biomarkers are critical to clinical decision-making in oncology. A 1991 NIH Consensus Conference (
1) stipulated that clinically useful prognostic and predictive factors in breast cancer must meet the following criteria:
They must provide significant, independent predictive value, validated by clinical testing.
Their identification and measurement must be feasible, reproducible, and widely available with quality control.
The results must be readily interpretable by clinicians and have therapeutic implications.
Measurement of biomarkers should not consume tissue needed for other tests, especially routine histopathological evaluation.
Prognostic markers provide information on the biological potential and most likely clinical course of a breast cancer irrespective of treatment (
2,
3). Insight into the natural history of individual breast cancers may provide valuable information regarding the need for systemic adjuvant therapy, but is uninformative with respect to which specific treatment regimen is most likely to be effective.
Predictive factors inform on the likelihood of response of a breast cancer to specific therapies (
3). Hormone receptor status predicts the responsiveness or lack of same of a breast cancer to endocrine therapy.
Some tumor biomarkers are of mixed significance. Estrogen receptor expression, while a strong predictor of response to endocrine therapy, is only weakly prognostic. HER2 expression has highly adverse prognostic implications but is predictive of tumor response to anti-HER2 therapy.
Conventional clinicopathological factors such as patient age, menopausal status, race/ethnicity, tumor size, nodal status, lymphovascular invasion, micrometastases or isolated tumor cells in regional lymph nodes, extracapsular extension of nodal metastases, tumor grade, tumor stage, presence or absence of the inflammatory phenotype, markers of tumor proliferation, and hormone receptor and HER2 status continue to be useful in estimating prognosis. The prognostic implications of the intrinsic breast cancer subtypes (
4,
5), multigene tumor signature assays (
6), and clinicopathological response to neoadjuvant systemic therapy (
7) offer further opportunities for refinement of clinical decision making in breast cancer.
Numerous potential breast cancer biomarkers have been cited and characterized over the past several decades. Discerning their true magnitude of effect, reliability and clinical utility has been complicated by deficiencies in biomarker assays and measurement, the quality of evidence supporting the potential biomarker status of the factor(s) under study, and failures in clinical trials design and studied patient cohorts and populations to account for and control confounding variables. A number of expert panels have reviewed available information on breast cancer biomarkers and concluded that limitations in available data allow for only the most guarded recommendations (
8). For these reasons, significant efforts have been directed toward standardizing the investigation and establishment of clinically relevant biomarkers.
GENERAL CONSIDERATIONS
For prognostic and predictive factors to be clinically useful, they must be detectable and reproducibly measurable by different laboratories at reasonable cost, yielding results promptly for clinical decision making. Their clinical correlations must be clearly defined in terms of their nature (prognostic, predictive, or both), and assay values, whether continuous or categorical, must be reliably associated with patient outcomes. The relevant clinical information being sought must not be available through another more readily accessible factor. The expected differences in outcomes must be significant and important from the patient’s perspective. Finally, useful prognostic and predictive factors must provide information upon which a choice among available treatment options can be based (
9).
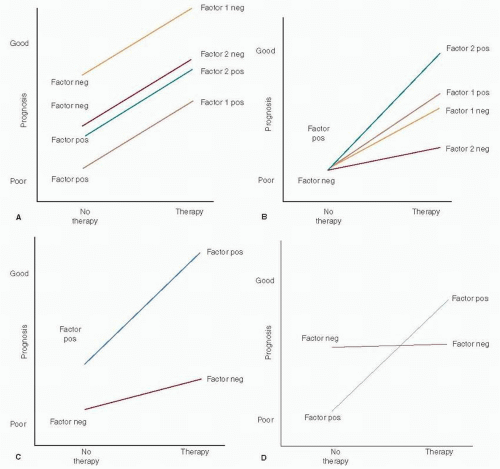
Pure prognostic and predictive factors are schematically depicted in
Figure 28-1, panels A and B, respectively.
Prognosis versus therapy are plotted as binomial variables (
8,
10). A large incremental difference in prognosis related to positive and negative status is observed for factor 1 (a strong prognostic factor such as lymph node status) while that for factor 2 is much smaller (ER status). In panel B, factor 1 is a weak predictive factor while factor 2 is a much stronger one. Hayes et al. (
10) proposed that prognostic factors in breast cancer be categorized quantitatively by their associated hazard ratios (HR), HR <1.5 denoting weak factors, 1.5 to 2.0 moderate factors, and >2.0 strong factors. They further proposed a similar rating of the strength of predictive factors by tumor response to and clinical benefit from a specific therapy. “Relative predictive value” (RPV), the ratio of the probability of response to treatment in a factor-positive patient as compared to that in a factor-negative patient, has been proposed as a means of quantifying the strength of predictive factors as weak (RPV = 1 — 2), moderate (RPV = 2 — 4), or strong (RPV > 4).
Panel C in
Figure 28-1 represents a mixed prognostic and predictive factor such as ER status, the prognostic effect being weakly favorable and the predictive effect strongly so. Panel D depicts a mixed factor with an unfavorable prognosis but highly responsive to specific therapy, as exemplified by HER2 status.
Statistical significance in marker-positive or marker-negative patient outcomes is not infrequently mistaken for or conflated as evidence of clinical utility. This does not always hold and should never be assumed. Along with the magnitude of the effect and the relevance of the marker, technical reliability and reproducibility are critically important, as is the design and execution of clinical studies (
10).
Technical shortcomings related to biomarker assay sensitivity, specificity, reproducibility, and reagent variability can be highly problematic. Standardization of assay methodologies has greatly improved, of late. For example, intra- and interobserver variation, well documented in immunohistochemical assays, has been controlled through automated and semiautomated processes (
8).
Determination of cut-off points that distinguish positive from negative results is critical to the development of clinically useful assays. Cut-off points can be set arbitrarily or based on data. Arbitrary cut-off point selection has included the limits of detection of the assay, two standard deviations above the normal mean, the mean value in affected as compared to normal patients, or an arbitrarily defined appropriate percentage of positive cells (
8).
Data-derived cut-off points have been based on plots of
p-values versus outcomes, plots of the magnitude of marker effect versus patient outcome, construction of receiver operating characteristic curves (cut-off points established by determining the optimal trade-offs of sensitivity and specificity in an assay), or subpopulation treatment effect pattern plot (STEPP) analysis (
11). The latter methodology evaluates outcomes to specific treatment regimens in subpopulations of patients within randomized trials or meta-analyses (
8).
Once established in a test group of patients, cut-off points must be confirmed in a validation patient cohort similar to but completely independent of the initial test group. Having been identified and validated, the clinical value of a new tumor marker relative to well-established prognostic or predictive factors is then confirmed by multivariate analysis. This provides information on the potential clinical utility of the new marker in medical practice.
Study design is key to identifying and establishing new tumor biomarkers. The Tumor Marker Utility Grading System (TMUGS) (
12) was developed as a frame of reference for grading the clinical utility of tumor markers based on published evidence. Putative markers are assigned a utility score according to degree of correlation with biological processes and end points (
Table 28-1) and favorable clinical outcomes (
Table 28-2), as determined by level of evidence (LOE;
Table 28-3) (
8) and grading of tumor marker studies (
Table 28-4) (
13).
LOE levels I and II are the most robust and objective, the ideal level I clinical trial being prospective, randomized, appropriately powered, and designed specifically to evaluate the clinical utility of a putative tumor marker for a discrete, predetermined use. That noted, a clinical trial adequately powered to determine a clinical end point may be underpowered for analysis of tumor marker subgroups by as much as 25%, even when tissue samples are available for all participating patients (
8).
Systematic overviews and pooled analyses of wellconducted LOE II studies are essentially equivalent to LOE I evidence. LOE III studies, with their greater variability in patient characteristics and therapies, are better suited to generating hypotheses than contributing clinically useful information (
8).
Attention to detail is critical to clinical trials design and conduct. The appropriate patient population must be selected with particular attention to a similar profile among them in terms of known prognostic factors. Trials focused on predictive factors are ideally prospective, randomized, and controlled, comparing patients receiving the intervention in question to untreated controls (
8).
The REMARK tool, a standardized reporting schema for tumor marker data, has been developed by the Working Group of the National Cancer Institute and the European Organization for Research and Treatment of Cancer (EORTC) (
14). This project was undertaken to eliminate the highly variable and flawed approaches to tumor marker elucidation and to provide a standard template for investigation of potential markers in the future.
 Breast Cancer Screening
Breast Cancer Screening
 Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
 Adjuvant Systemic Therapy: Endocrine Therapy
Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Preoperative Endocrine Therapy for Operable Breast Cancer



