Abstract
Acute myeloid leukaemia (AML) is a highly complex and heterogeneous disease. Proper classification according to the 2016 World Health Organization (WHO) classification requires a systematic approach and integration of key clinical, laboratory, pathologic and genetic information [1, 2]. Great advances in our understanding of the pathogenesis and molecular underpinnings of AML have been realized since the original AML classification using the French–American–British (FAB) system (1976). This genetic revolution not only contributes to enhanced disease diagnosis and prognostication but also to ongoing improvements in therapeutic strategies.
Introduction
Acute myeloid leukaemia (AML) is a highly complex and heterogeneous disease. Proper classification according to the 2016 World Health Organization (WHO) classification requires a systematic approach and integration of key clinical, laboratory, pathologic and genetic information [1, 2]. Great advances in our understanding of the pathogenesis and molecular underpinnings of AML have been realized since the original AML classification using the French–American–British (FAB) system (1976). This genetic revolution not only contributes to enhanced disease diagnosis and prognostication but also to ongoing improvements in therapeutic strategies.
This chapter focuses on the four broad AML categories recognized by the WHO classification, (AML with recurrent genetic abnormalities, AML with myelodysplasia-related changes, therapy-related myeloid neoplasm and AML, not otherwise specified) [3–6]. We review the key morphologic, immunophenotypic and genetic features of each group and relevant subentities. Myeloid sarcoma and myeloid proliferations associated with Down syndrome are also briefly discussed.
Acute Myeloid Leukaemia Classification
The 2016 WHO classification recognizes 25 subtypes of AML and related precursor neoplasms (Table 9.1) [1–6]. Compared to the 2008 WHO classification, the 2016 classification scheme includes an expanded number of entities with recurring genetic abnormalities, including two provisional entities (AML with RUNX1 mutation and AML with BCR-ABL1), refinements of diagnostic criteria and gene nomenclature, and relocation of the entity blastic plasmacytoid dendritic cell neoplasm. The category of AML with myelodysplasia-related changes has several important changes including a refinement of the myelodysplasia-associated cytogenetic abnormalities and clarification of the importance of recurring genetic abnormalities over pure morphologic dysplasia. Therapy-related AML remains as an important biologic category. AML, not otherwise specified, consists of a heterogeneous set of entities with variable prognosis and which do not meet criteria for any of the aforementioned categories.
| AML with recurrent genetic abnormalities | |
| AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1 | |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22);CBFB-MYH11 | |
| Acute promyelocytic leukaemia (APL) with PML-RARA | |
| AML with t(9;11)(p21.3;q23.3); KMT2A-MLLT3 | |
| AML with t(6;9)(p23;q34.1);DEK-NUP214 | |
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM | |
| AML (megakaryoblastic) with t(1;22)(p13.3;q13.1);RBM15-MKL1 | |
| Provisional entity: AML with BCR-ABL1 | |
| AML with mutated NPM1 | |
| AML with biallelic mutations of CEBPA | |
| Provisional entity: AML with mutated RUNX1 | |
| AML with myelodysplasia-related changes | |
| Therapy-related myeloid neoplasms | |
| AML, not otherwise specified | |
| AML with minimal differentiation | |
| AML without maturation | |
| AML with maturation | |
| Acute myelomonocytic leukaemia | |
| Acute monoblastic/monocytic leukaemia | |
| Pure erythroid leukaemia | |
| Acute megakaryoblastic leukaemia | |
| Acute basophilic leukaemia | |
| Acute panmyelosis with myelofibrosis | |
| Myeloid sarcoma | |
| Myeloid proliferations associated with Down syndrome | |
| Transient abnormal myelopoiesis (TAM) | |
| Myeloid leukaemia associated with Down syndrome | |
Acute Myeloid Leukaemia Diagnostic Approach
The successful and correct diagnosis of AML requires a systematic and integrated approach. A logical, systematic approach includes assessment of the clinical history, complete blood cell count (CBC), blast identification and enumeration, morphologic assessment of dysplasia, immunophenotypic studies and genetic testing (Table 9.2). Integration of these factors will allow for proper classification and prognostication of AML cases. For example, refinement of a final diagnosis of AML to therapy-related AML or AML with myelodysplasia-related changes is possible based on clinical history of prior toxic chemotherapy/radiotherapy or known prior myelodysplastic syndrome, respectively.
Table 9.2 Systematic approach to the diagnosis of acute myeloid leukaemia.
| Feature | Comments |
|---|---|
| Clinical data | History of chemotherapy/radiotherapy; history of MDS or MDS/MPN |
| CBC | Haematopoietic failure (markedly reduced RBC, absolute neutrophil and platelet counts) |
| Blast identification | Recognize blasts and blast equivalents (see Table 9.3) |
| Blast enumeration | Blasts ≥20% of peripheral blood and/or bone marrow cellsa |
| Promonocytes always included in blast percentage | |
| Promyelocytes only included in blast percentage in APL | |
| Blast percentage based on total bone marrow cells for all AML subtypes | |
| Proerythroblasts included in determining blast percent only in PEL | |
| Blast enumeration based on morphologic differential cell count (not flow cytometry percentage) | |
| Cytochemical stains | Myeloperoxidase (MPO) most specific cytochemical stain for granulocyte precursors; May be weakly positive in monocyte precursors but generally negative |
| Non-specific esterase (alpha-naphthyl butyrate esterase) shows diffuse positivity of monocytic cells (reaction may be weak to strong) | |
| Immunophenotypic studies | Confirm myeloid lineage (see Table 9.3) |
| Cytogenetics | Perform in all cases; AML-defining versus other |
| FISH | Unnecessary in most cases; useful for STAT confirmation of PML-RARA in APL; may be useful in cases with monocytic differentiation, which may have cryptic rearrangements of KMT2A (previously known as MLL) and CBFB-MYH11 |
| Molecular genetics | FLT3, CEBPA, NPM1, RUNX1, KIT, BCR-ABL1, others |
a Blasts may be less than 20% in cases with t(8;21), inv(16) and PML-RARA; different diagnostic criteria for erythroblasts in pure erythroid leukaemia.
Key: CBC, complete blood cell count; MDS, myelodysplastic syndrome; MDS/MPN, myelodysplastic/myeloproliferative syndrome; APL, acute promyelocytic leukaemia; FISH, fluorescence in situ hybridization; PEL, pure erythroid leukaemia.
Blast identification and enumeration is of fundamental importance in establishing an AML diagnosis. A diagnosis of AML requires the presence of ≥20% blasts in most cases. The exceptions include AML with t(8;21), inv(16)/t(16;16) and acute promyelocytic leukaemia (APL) with PML-RARA, in which the diagnosis may be established without the requisite 20% blasts. The gold standard for determining the blast percentage is by differential count of nucleated cells in the peripheral blood and/or bone marrow (BM). Table 9.3 highlights the different types of blasts and blast equivalents, their typical cytologic characteristics and their immunophenotypic profiles.
Table 9.3 Blast identification, cytologic characteristics and typical immunophenotypic (IP) features.
| Blast type | Cytologic characteristics | Usual IP profile |
|---|---|---|
| Myeloblast | Large nucleus with finely dispersed chromatin, variably prominent nucleoli; variable number cytoplasmic granules | CD34, HLA-DR, CD13, CD33, CD117 MPO, weak CD64, weak CD4, weak CD45 |
| Promyelocytea | Nuclear folding and lobulation characteristic of microgranular variant of APL; may see intense cytoplasmic granularity dispersed throughout cytoplasm | CD13, CD33, CD15, MPO, CD45, typically CD34 and HLA-DR negative |
| Monoblast | Nuclear chromatin finely dispersed with variably prominent nucleoli; nuclei round to folded; abundant, lightly basophilic cytoplasm containing fine granulation and occasional vacuoles; low nuclear to cytoplasmic ratio | HLA-DR, CD13, CD33, bright CD36/CD64, CD34 and CD117 often negative |
| Promonocyte | Slightly condensed yet immature nuclear chromatin; delicate nuclear folds and grooves; variably prominent nucleoli; abundant blue/grey cytoplasm that may be vacuolated with fine azurophilic granules | HLA-DR, CD13, CD33, bright CD36/CD64, CD34 and CD117 negative |
| Erythroblastb | Round nucleus with reticulated chromatin; nucleoli variably prominent; moderate amounts of deeply basophilic cytoplasm that may be vacuolated | CD71, CD117, E-Cadherin, variable/often negative for glycophorin A and haemoglobin A, typically CD45 and CD34 negative |
| Megakaryoblast | Often not recognizable without special studies, may be lymphoid-appearing with high nuclear to cytoplasmic ratio; nuclear chromatin fine to variably condensed; cytoplasm usually agranular or with few granules; cytoplasmic blebs may be seen but not specific | HLA-DR, CD41, CD13, variable CD33, CD61, variable CD34, CD31, often CD56, often CD7, variable CD45 |
a Blast equivalent in APL; bBlast equivalent in PEL
Key: APL, acute promyelocytic leukaemia; PEL, pure erythroid leukaemia.
Key Components of the Initial Diagnostic Work-Up of AML
Given the importance of and need for an accurate AML diagnosis, multiple guidelines driven by expert consensus, data and peer-reviewed published literature now exist [7–9]. Such guidelines highlight the key testing that should be considered in all newly diagnosed cases of AML. Such testing should include relevant clinical data, current CBC and peripheral blood smear review, fresh BM aspirate for morphologic review, comprehensive flow cytometric immunophenotyping, conventional cytogenetic analysis and molecular genetic testing (DNA and/or RNA) (Table 9.4) In adult patients with AML, FLT3 ITD testing should be performed regardless of karyotype and additional testing for KIT mutations is needed in core-binding factor AML [inv(16) and t(8;21)]. For patients without core-binding factor AML, APL or AML-MRC with the associated genetic abnormalities, assessment for NPM1, RUNX1 and CEBPA mutations should be pursued. With improved access to large next-generation sequencing panels as well as development of targeted therapies, testing for additional gene mutations, including ASXL1, FLT3 TKD, DNMT3A, IDH1, IDH2, TET2, WT1 and TP53 may also be warranted.
| Component | Comment(s) |
|---|---|
| Clinical data | Pertinent cytotoxic history, history of prior myeloid disorder |
| CBC | Recently performed; peripheral blood smear morphologic review |
| Bone marrow aspirate | Fresh for morphologic review, flow cytometry, conventional cytogenetics and molecular genetics (DNA and/or RNA) |
| Cytogenetics | Perform in all cases |
| FLT3-ITD | Perform in all cases |
| KIT mutation | Perform in core-binding factor AML; inv(16) and t(8;21) |
| NPM1, RUNX1 and CEBPA mutations | Perform in all cases that do not meet criteria for core-binding factor AML, APL and AML-MRC (based on history or karyotype) |
| FISH | Useful in selected cases to confirm suspected cytogenetic abnormality |
| DNA extract and hold | Useful in situations with challenging operational logistics to ensure that if certain testing is needed, there is available specimen to perform |
| Final report | Utilize 2016 WHO terminology |
Key: FISH, fluorescence in situ hybridization; CBC, complete blood cell count; ITD, internal tandem duplication; APL, acute promyelocytic leukaemia; AML-MRC, acute myeloid leukaemia with myelodysplasia-related changes
Acute Myeloid Leukaemia Diagnostic Categories
AML with Recurrent Genetic Abnormalities
The 11 subtypes of leukaemia in this category have characteristic morphologic, clinical and prognostic features in addition to specific cytogenetic and molecular genetic findings (Table 9.5.) The 2016 WHO classification introduces two new provisional entities (AML with BCR-ABL1 and AML with mutated RUNX1), codifies two previous provisional entities to bona fide entities, AML with mutated NPM1, and AML with biallelic mutations of CEBPA (the diagnostic criteria are refined to emphasize that the favourable prognosis is attributable to biallelic mutations), modifies the gene nomenclature of MLL to KMT2A, refines the inv(3) nomenclature to indicate that GATA2 and MECOM are not fused, and removes the cytogenetic translocation from APL (Table 9.1). Given the unique clinicopathologic and genetic features of each of these entities, they are discussed briefly in the following paragraphs and key attributes are highlighted in Table 9.5.
AML with t(8;21)(q22;q22.1); RUNX1–RUNX1T1
This AML is common in both the paediatric and adult populations, demonstrates characteristic cytologic features, has a unique immunophenotypic profile and is associated with an overall favourable prognosis. Importantly, the presence of this genetic abnormality is diagnostic of AML regardless of the blast count [3, 10, 11]. This AML subtype, along with AML with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22); CBFB-MYH11, both involve genes in the core-binding factor complex and are thus referred to collectively as core-binding factor AMLs.
Table 9.5 Key clinicopathologic attributes of the subtypes of AML with recurring genetic abnormalities.
| Diagnostic entity | Features |
|---|---|
| AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 | Common in paediatric and adult populations; classic blast appearance with cytoplasmic hofs, single and thin Auer rods, basophilic cytoplasm and abundant pink granules |
| Background neutrophilic dysplasia is not uncommon | |
| Blasts express typical myeloid markers but also B-associated markers as well (CD19, PAX5, cytoplasmic CD79a); should not be misdiagnosed as mixed phenotype acute leukaemia (B/myeloid) | |
| The requisite 20% blasts for a diagnosis of AML is not required | |
| Overall favourable prognosis | |
| KIT mutation should be assessed; may be associated with worse prognosis in adults | |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 | Common in paediatric and adult populations |
| AML with monocytic differentiation and abnormal eosinophils (dark, chunky granules) | |
| Blasts are an admixture of myeloid and monocytic blasts often with a background of maturing monocytic and granulocytic components | |
| The requisite 20% blasts for a diagnosis of AML is not required | |
| Overall favourable prognosis | |
| KIT mutation should be assessed; may be associated with worse prognosis in adults | |
| Has an increased propensity for extramedullary disease involvement (e.g. CNS, intestine) | |
| Acute promyelocytic leukaemia (APL) with PML-RARA | Common in children and middle-age adults |
| Prompt recognition is essential due to potential life-threatening coagulopathy | |
| Morphologic differentiation of blasts with tretinoin therapy | |
| Two morphologic variants: hypergranular and hypogranular | |
| Abnormal promyelocytes of the hypergranular variant are considered blast equivalents for the purpose of establishing the AML diagnosis | |
| Hypergranular variant immunophenotype typically CD117, bright CD33, strong MPO positive with lack of HLA-DR and CD34; microgranular variant often shows dim to moderate CD34 and/or HLA-DR | |
| Overall favourable prognosis | |
| FLT3 mutations may be present; FLT3-ITD may be associated with the microgranular morphologic variant | |
| AML with t(9;11)(p21.3;q23.3); KMT2A- MLLT3 | Common in the paediatric population |
| KMT2A formerly known as MLL | |
| Typically monocytic/monoblastic features | |
| Extramedullary disease involvement not unexpected | |
| Usual monocytic phenotype: bright co-expression of CD36/CD64, CD33, weak CD4 and often CD34 and CD117 negative | |
| Intermediate prognosis | |
| AML with t(6;9)(p23;q34.1); DEK-NUP214 | Rare subtype of AML |
| Multilineage dysplasia and basophilia are characteristic | |
| Monocytic features and Auer rods may be seen | |
| Typical myeloid blast phenotype | |
| FLT3-ITD mutations are very common | |
| Poor prognosis | |
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM | Relatively uncommon (1% of AML cases) |
| Fairly distinctive morphology: often thrombocytosis with dysplastic features, increased and markedly dysplastic megakaryocytes and multilineage dysplasia including pseudo-Pelger–Huet cells | |
| Immunophenotype of blasts is typical myeloid: CD34, CD117, CD13 and CD33; may see aberrant CD7 | |
| Often additional cytogenetic aberrations (e.g. –7, del(5q) and complex karyotype) | |
| Mutations in NRAS, PTPN11, FLT3 and/or KRAS are common | |
| Poor prognosis | |
| AML (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1 | Very uncommon AML |
| Typical infantile presentation with hepatosplenomegaly | |
| Megakaryoblasts express one or more of CD41, CD42b, CD61: most often CD34, HLA-DR, MPO and CD45 negative | |
| Significant myelofibrosis in the core biopsy; may preclude an optimal aspiration | |
| High-risk disease | |
| Provisional entity: AML with BCR-ABL1 | Rare subtype of de novo AML |
| Must unequivocally exclude prior/subsequent chronic myeloid leukaemia | |
| Compared with chronic myeloid leukaemia, this entity exhibits less frequent splenomegaly, insignificant basophilia, lower bone marrow cellularity (80% versus 95–100%) and micromegakaryocytes are uncommon | |
| Monosomy 7 and trisomy 8 may be seen in addition to the BCR-ABL1 abnormality | |
| Prognosis appears adverse | |
| AML with mutated NPM1 | Common AML occurring in 5–9% of children and one third of adults |
| Female predominance | |
| Majority of cases show monocytic differentiation | |
| Mutation detected by NPM1 immunohistochemistry (aberrant cytoplasmic localization) or DNA sequencing | |
| Prognosis depends on the presence (or not) of FLT3-ITD and should be performed in all cases | |
| Prognosis is favourable for NPM1 mutated/FLT3-ITD wild-type | |
| AML with biallelic mutations of CEBPA | Present in ~5–10% of children and younger adults |
| No diagnostic unique morphologic features | |
| Blasts more often express HLA-DR, CD7 and CD15 | |
| Favourable prognosis | |
| Work-up should include evaluation for an underlying germline predisposition syndrome | |
| Karyotype most often normal | |
| Provisional entity: AML with mutated RUNX1 | Must exclude other AML diagnostic categories |
| Not uncommon (10% of AML cases) | |
| Often exhibits features of AML with minimal differentiation | |
| No unique immunophenotypic features | |
| Typically monoallelic RUNX1 mutation | |
| Prognosis appears adverse |
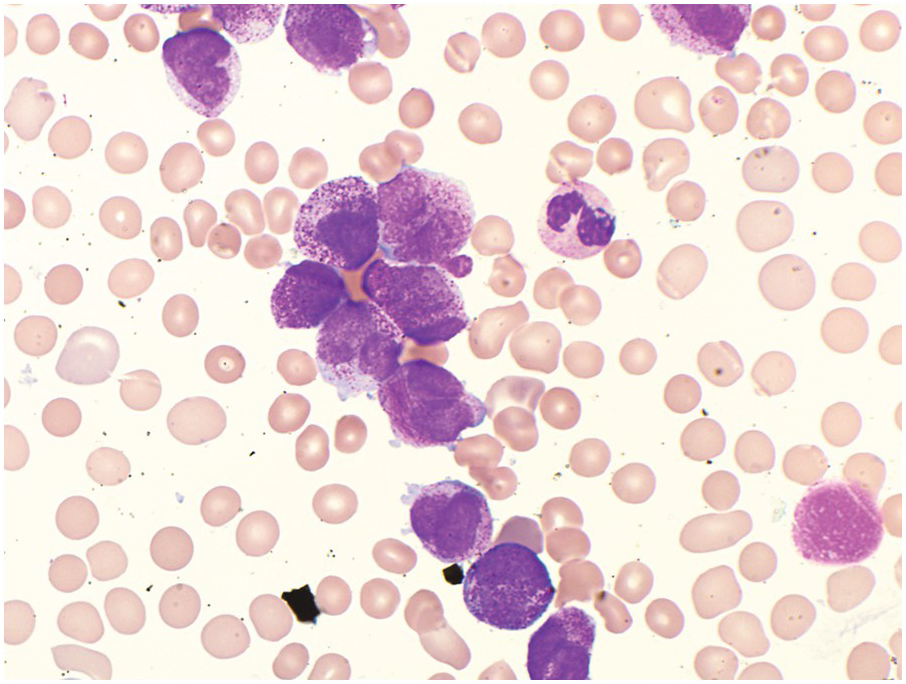
AML with t(8;21) typically shows myeloid blasts with abundant basophilic cytoplasm, prominent cytoplasmic hofs, salmon-coloured granulation, sometimes chunky, pseudo-Chediak–Higaski type granules and single, thin Auer rods with tapered ends in a background of neutrophilic maturation with variable dysplastic features (Figure 9.1). Dysplasia is absent for the most part in the erythroid and megakaryocytic lineages. A monocytic component tends to be inconspicuous. Immunophenotypically, AML with t(8;21) expresses typical blast and myeloid markers CD34, CD13, CD33, myeloperoxidase (MPO) and TdT, and also shows aberrant expression of B-lineage associated markers, CD19, PAX5 and CD79a. The presence of B-lineage markers does not indicate a diagnosis of mixed phenotype acute leukaemia (B/myeloid type).
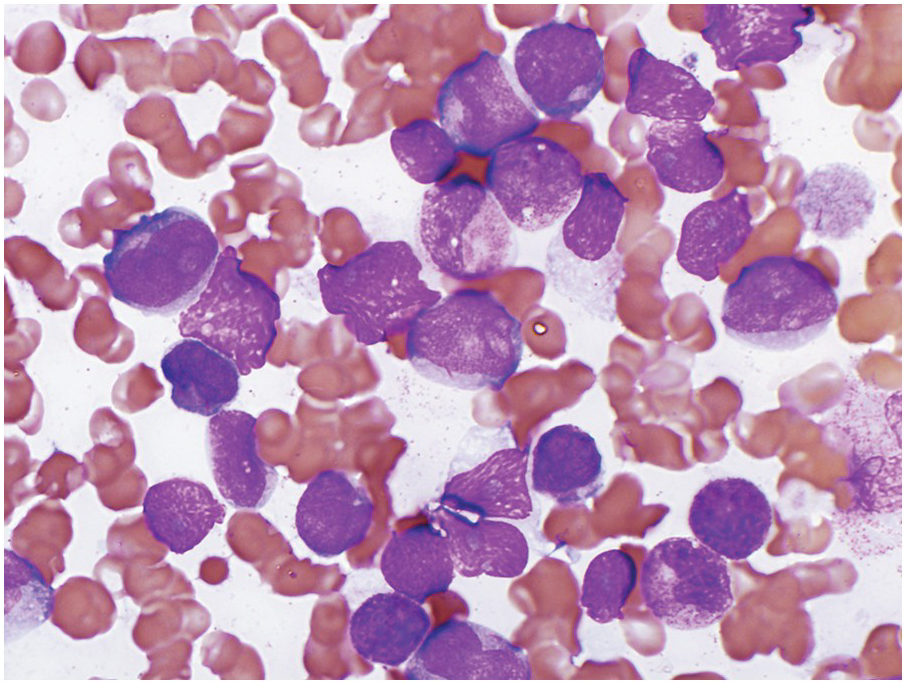
Figure 9.1 AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1. Blasts show distinct perinuclear hofs and salmon-coloured cytoplasmic granules. Occasional blasts containing single, thin Auer rods are also present.
AML with t(8;21) is associated with an overall favourable prognosis in children and adults, especially when treated with high-dose cytarabine. Mutations of KIT in core-binding factor AML are common (~25%) [12, 13, 14]. In adults, KIT mutations appear to worsen prognosis. It is unclear if they have a similar prognostic effect in children.
Acute Myeloid Leukaemia with inv(16)(p13.1;q22) or t(16;16)(p13.1;q22); CBFB-MYH11
This AML, similar to AML with t(8;21), is common in both the paediatric and adult populations, demonstrates characteristic cytologic features, is associated with an overall favourable prognosis and may be diagnosed with this genetic finding as AML regardless of the blast count [3, 10, 11]. AML with inv(16) has a characteristic morphology of an acute myeloid leukaemia with monocytic differentiation accompanied by a unique increase in eosinophils with abnormal morphology in the BM (Figure 9.2). Previously, in the FAB classification, this AML was referred to as AML M4 with abnormal eosinophils (M4Eo). The blastic component consists of an admixture of myeloblasts, monoblasts and promonocytes. The abnormal eosinophils have abundant and large basophilic granules. Importantly, while most of these cell types are all typically seen in the peripheral blood, the increased and abnormal-appearing eosinophils are largely restricted to the BM. Therefore when reviewing the peripheral blood smear one needs to have a high index of suspicion for this entity and strongly suggest BM evaluation.
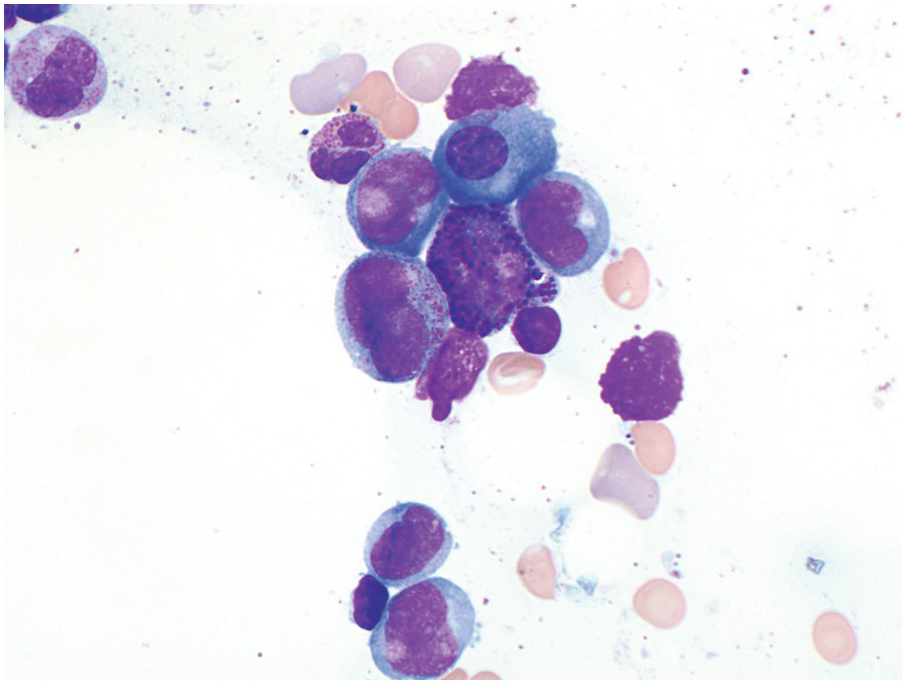
Immunophenotypic assessment most often demonstrates multiple abnormal populations including typical myeloid blasts (CD34, CD117, C13 and/or CD33 and MPO positive), a monocytic population (CD36/CD64 bright positive, CD14, weak CD4, lysozyme) and a background granulocytic population expressing myeloid markers [15, 16]. Cytochemical staining for NSE is useful in highlighting the monocytic component.
Cytogenetically this genetic abnormality may be subtle and/or cryptic, therefore notification of the genetic laboratory to the possibility of this subtype of AML is critical. FISH studies and/or molecular assessment of CBFB-MYH11 transcripts may also be useful such that this entity, which is associated with an overall favourable prognosis, is not overlooked. Additional cytogenetic abnormalities may be seen in a subset of cases, particularly trisomy 22 as well as trisomy 8.
AML with inv(16) is associated with an overall favourable prognosis. Similar to AML with t(8;21), KIT mutations are present in approximately 30% of cases and may negatively impact on the prognosis in the adult population [18].
Acute Promyelocytic Leukaemia with PML-RARA
Acute promyelocytic leukaemia (APL) with PML-RARA is a relatively common AML primarily affecting the younger and middle-age populations. This diagnosis is absolutely critical and should be promptly recognized because of the attendant risk of life-threatening disseminated intravascular coagulation and associated significant morbidity and mortality. If this diagnosis is suspected, rapid FISH and/or molecular testing should be performed. The clinical service should be immediately notified such that appropriate testing and interventions may begin.
The genetic alteration (PML-RARA) is the result of the t(15;17) with fusion of the promyelocytic leukaemia gene (PML) on chromosome 15 with the retinoic acid receptor (RARA) gene on chromosome 17. Detection of this abnormality is diagnostic of APL regardless of the blast count. The PML-RARA fusion protein mediates a block in myeloid differentiation, which can be overcome using all-trans-retinoic acid treatment (tretinoin, previously known as ATRA) therapy and arsenic trioxide. The blasts differentiate in response to tretinoin. In many instances of standard risk APL, tretinoin and arsenic trioxide achieve adequate remission [17]. In high-risk patients, however, the addition of anthracyclines is needed for long-term remission.
Two morphologic variants, hypergranular and hypogranular, are recognized and have distinct morphologic and immunohistochemical features [3, 10]. The hypergranular variant of APL comprises the majority of cases (70%) and generally presents with leukopaenia. Meticulous assessment of the feather edge and sides of the peripheral blood smear may be the first clue to this critical diagnosis. The abnormal promyelocytes have numerous large red to purple cytoplasmic granules, often obscuring the nuclear contours (Figure 9.3). Most cases have cells that contain numerous Auer rods. The abnormal promyelocytes are considered blast equivalents for the purpose of diagnosing APL because usual myeloblasts may not be increased. Immunophenotyping of the abnormal promyelocytes typically shows increased side-angle light scatter properties, expression of CD117, bright CD33, strong MPO, and the characteristic absence of HLA-DR and CD34 [3, 18, 19]. It is important to note that this characteristic combined CD34 and HLA-DR negative immunophenotype is not specific to APL and may be observed in other AML cases including those that are cytogenetically normal AML with cup-like nuclear invaginations.