Abstract
Thrombocytopenia occurs in less than 1% of all newborns. However, thrombocytopenia is a common finding in the intensive-care nursery where it is present in 25–35% of admitted infants [1, 2]. See Chapters 12 and 13 for a discussion of the approach to thrombocytopenia as well as acquired causes of thrombocytopenia in newborns. This chapter will focus primarily on the diagnosis and initial management of inherited thrombocytopenia disorders that present in infancy.
Approach to Thrombocytopenia in the Newborn
Thrombocytopenia occurs in less than 1% of all newborns. However, thrombocytopenia is a common finding in the intensive-care nursery where it is present in 25%–35% of admitted infants [1, 2]. See Chapters 12 and 13 for a discussion of the approach to thrombocytopenia as well as acquired causes of thrombocytopenia in newborns. This chapter will focus primarily on the diagnosis and initial management of inherited thrombocytopenia disorders that present in infancy.
Acquired causes of thrombocytopenia are much more common than inherited ones at any age, and thus it can be challenging to recognize the rare situations when thrombocytopenia has a genetic basis. In general, an inherited condition can be suspected in newborns or infants if thrombocytopenia is persistent and unexplained, if there are characteristic findings on the peripheral blood smear, if there are associated congenital abnormalities, or if there is a family history suggestive of a thrombocytopenia syndrome. There are special challenges, however, in trying to identify inherited causes of thrombocytopenia in neonates. First, despite their individual rarity, there is a large number of genetic disorders associated with thrombocytopenia. Mutations in more than 30 different genes have been described in inherited thrombocytopenia syndromes [3]. Diagnosis can be challenging as the phenotype associated with a given mutant gene can vary from patient to patient, and in some disorders characteristic findings may not present until later in life. Laboratory studies can also be difficult to interpret in a young child. Although review of the peripheral blood smear is of key importance in evaluating patients with thrombocytopenia, there are important caveats to consider when looking at a smear from a newborn. Acquired conditions that lead to increased platelet turnover can result in the finding of large platelets, and white cell inclusions may be seen in the setting of infection. Platelet clumping is a frequent artifact when blood is obtained from a heel stick. Normal ranges for measures such as the red blood cell MCV are higher in newborns than in older infants and adults, and von Willebrand antigen levels are increased due to the influence of maternal hormones. Time may be required to determine if specific findings are persistent. From a practical standpoint, it can be difficult to do an extensive workup on a small baby, due to either limitations in drawing blood or sedation risks, and in some cases it might be reasonable to either delay the evaluation or to try to establish the diagnosis from an affected parent. Finally, the use of genetic testing in young children is controversial: In general, if it will not affect clinical management of the child then one should consider delaying genetic testing until the child is old enough to understand its implications (A. Greinacher, personal communication). On the other hand, a timely genetic diagnosis may be important for counseling the parents about their risk of having another affected child; and may have implications for other family members. It is important that the family receive appropriate genetic counseling, particularly when considering diagnoses that include a risk for cancer. For these reasons and others, it may not be possible to establish a diagnosis of inherited thrombocytopenia in neonates. To allow a broader discussion, this chapter will consider thrombocytopenia in the newborn period and extending through infancy.
With these caveats in mind, the approach to the diagnosis of inherited thrombocytopenia should begin with a review of the peripheral blood smear as well as a careful history and physical exam. These evaluations are readily available and can suggest the diagnosis and inform the subsequent workup.
Review of the Peripheral Smear
Review of the peripheral smear is an essential part of any thrombocytopenia evaluation, in particular noting platelet size and granularity, red cell morphology, and the presence or absence of white cell inclusions [4]. Inherited thrombocytopenia syndromes are classified by platelet size [5]. To gauge size on the peripheral smear, platelets can be compared to the diameter of a red blood cell. Platelets that are more than 50% of the diameter of a red cell are considered large, whereas platelets that are greater than the diameter of a red cell are referred to as giant. The mean platelet volume (MPV) reported on most automated cell analyzers is another readily available measure of platelet size, with the caveat that platelets that are very large or very small may be excluded from the analysis. If there are some large or giant platelets present on a background of small or normal sized platelets, this suggests a state of increased platelet turnover such as is seen in immune thrombocytopenia, type 2B von Willebrand disease or thrombotic thrombocytopenic purpura. A pale or “gray” appearance to the platelets may indicate a deficiency in alpha granules. Platelet clumping may be an artifact, but it is also characteristic of von Willebrand disease type 2B. Red cell polychromasia suggests hemolysis. The presence of schistocytes suggests that hemolysis is due to a microangiopathic process such as thrombotic thrombocytopenic purpura, whereas stomatocytes should trigger consideration of sitosterolemia. Macrocytosis can be seen in the setting of underlying marrow failure syndromes such as Fanconi anemia, Diamond–Blackfan anemia or dyskeratosis congenita. Granulocytes should be carefully examined for the presence of Dohle-like inclusion bodies that are diagnostic of MYH9-related disorder. Representative peripheral smear findings are shown in Fig. 15.1.
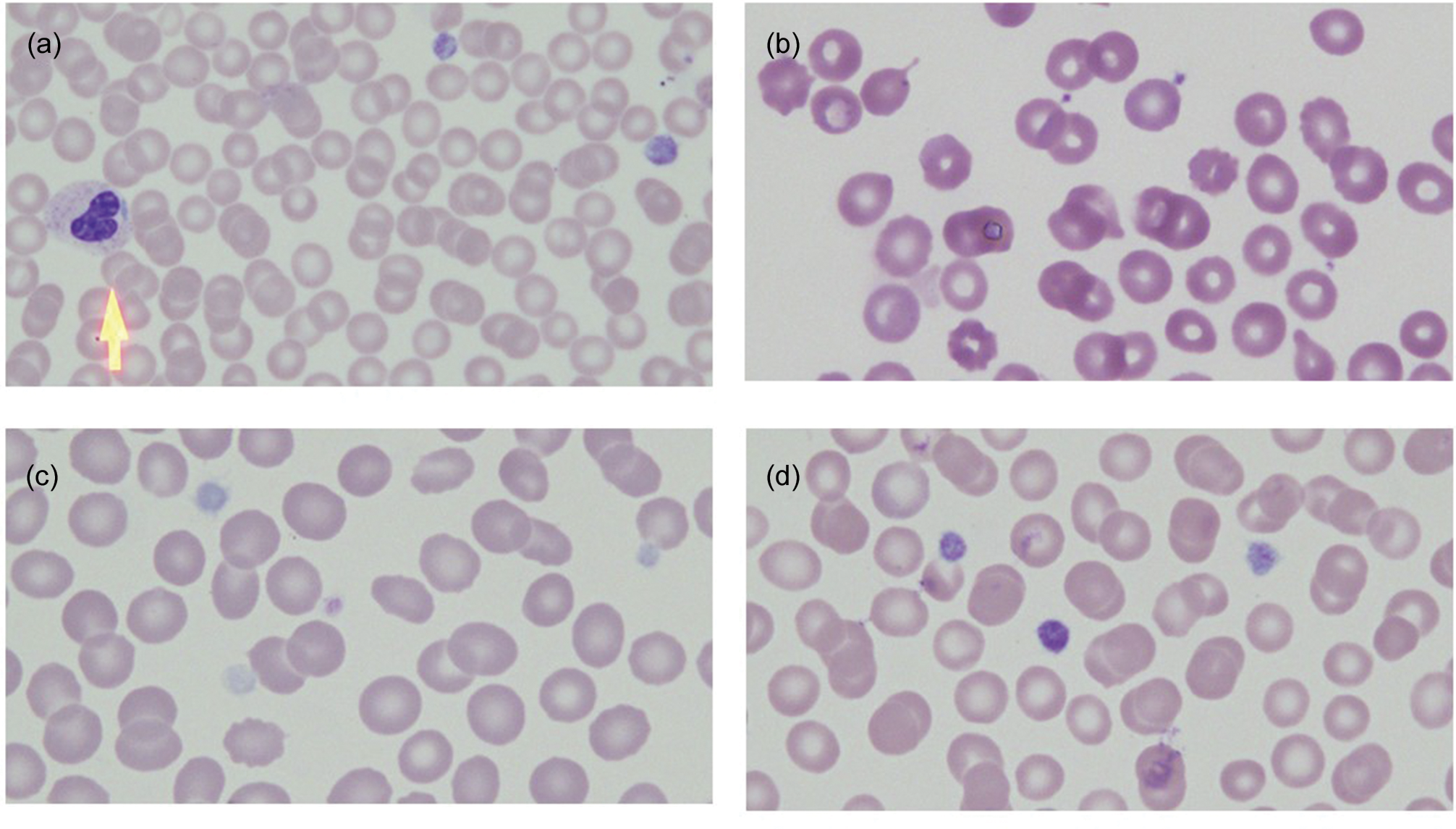
Fig. 15.1 Peripheral smear findings in representative inherited thrombocytopenia syndromes. (a) MYH9-related disorder. Arrow indicates neutrophil with a Dohle-like inclusion. (b) Small platelets in Wiskott–Aldrich syndrome. (c) Pale platelets in Gray platelet syndrome. (d) Large platelets in Bernard–Soulier syndrome. Magnification 100×.
Family History
A careful family history may establish the inheritance pattern as autosomal dominant, autosomal recessive, X-linked, or sporadic. If the parents are related or if they are from similar ethnic or religious backgrounds, the chances of an autosomal recessive condition are increased. Thrombocytopenia that is asymptomatic may be unrecognized, so it cannot always be assumed that a family member is unaffected if they have not been tested. In addition, some thrombocytopenia syndromes include a predisposition to complications that may not manifest until later in childhood or in adulthood, such as bone marrow failure, myelodysplasia or leukemia, renal failure, deafness, pulmonary or hepatic fibrosis, or other cancers. A detailed history that includes three generations may provide important clues to the diagnosis.
Exam Findings
While congenital abnormalities do not always accompany inherited thrombocytopenia syndromes, if present they may help to establish the diagnosis. It is important to examine the forearms carefully. Bilateral radial hypoplasia or aplasia is a feature of thrombocytopenia with absent radii. If thumbs are missing or otherwise abnormal, then Fanconi anemia should be considered. Radioulnar synostosis, which can be detected by the limitation of rotational movement of the forearm, has been associated with mutations in several genes, including MECOM and HOXA11. Plain X-rays of the forearms can be useful to detect subtle abnormalities. Additional anomalies, such as poor growth, renal or cardiac malformations, developmental delay, or hearing loss, may indicate the presence of a congenital syndrome associated with thrombocytopenia. Congenital abnormalities are frequent in inherited bone marrow failure syndromes such as Diamond–Blackfan anemia and Fanconi anemia. While Diamond–Blackfan anemia is classically thought to be a “pure red cell aplasia,” thrombocytopenia or neutropenia may be present or even dominate the clinical picture, particularly in the newborn period [6]. Similarly, low platelets may be the first cytopenia to be recognized in children with Fanconi anemia. Thrombocytopenia is common in a range of other congenital disorders such as Paris-Trousseau syndrome, Noonan syndrome, del22q11.2 syndrome and trisomy 13, 18 or 21. In contrast, other inherited platelet disorders, such as congenital amegakaryocytic thrombocytopenia, are not associated with congenital abnormalities and may be initially difficult to differentiate from acquired causes of low platelets such as auto- or alloimmune thrombocytopenia.
Subsequent Evaluations and Supportive Care
Depending on the suspected diagnosis, additional evaluations may be appropriate (see Table 15.1).
| Study | Useful to assess | Diagnostic implications |
|---|---|---|
| Complete blood count | Blood counts and parameters | All |
| Reticulocyte count | Red cell production | DBA, TTP |
| Immature platelet fraction | Platelet production | VWD2B, TTP |
| Peripheral smear | Morphology of blood cells | All |
| Bone marrow aspiration and biopsy | Marrow cellularity, megakaryocyte number, dysplasia, fibrosis, storage disorder | CAMT, RUSAT, FA and DC, GPS, RUNX1, ANKRD26, ETV6 |
| Platelet aggregation | Platelet function | BSS, GT |
| Platelet electron microscopy | Platelet granules | GPS, PTS |
| Immunologic studies | Hypogammaglobulinemia, lymphopenia | WAS, DC, 22q11.2 deletion, RUSAT |
| Chromosomal breakage studies | DNA repair defect | FA |
| Cytogenetic studies | Numerical abnormalities, deletions or translocations of chromosomes | Trisomies, 22q11.2 deletion, PTS |
| von Willebrand assays | vWF activity, multimers | VWD2B, TTP |
| ADAMTS13 activity | Microangiopathic hemolytic anemia | TTP |
| Forearm xrays | Radial hypoplasia, radioulnar synostosis | TAR, RUSAT, FA |
| Echocardiogram | Congenital heart defects | FA, TAR, DBA, 22q11.2 del, PTS, NS |
| Abdominal US | Renal abnormalities | FA, DBA, TAR |
| Brain MRI | Cerebellar hypoplasia | DC/HHS |
| Genetic studies | Specific mutation analysis | All |
Abbreviations: DBA (Diamond–Blackfan anemia), TTP (thrombotic thrombocytopenic purpura), vWD (von Willebrand disease), CAMT (congenital amegakaryocytic thrombocytopenia), RUSAT (radioulnar synostosis with amegakaryocytic thrombocytopenia), FA (Fanconi anemia), DC (dyskeratosis congenita), GPS (gray platelet syndrome), BSS (Bernard–Soulier syndrome), GT (Glanzmann thrombasthenia), PTS (Paris-Trousseau syndrome), WAS (Wiskott–Aldrich syndrome), NS (Noonan syndrome), HHS (Hoyeraal–Hreidarsson syndrome).
Tools for the management of bleeding symptoms in children with inherited thrombocytopenia include platelet transfusions and antifibrinolytics. In infants, platelets are dosed at 10–15 milliliters per kilogram. Standard recommendations dictate that all blood products should be leukocyte reduced; leukocyte reduction minimizes the occurrence of febrile reactions, prevents transmission of CMV, and reduces alloimmunization. Irradiation of blood products should be performed if the donor is closely related, or if the recipient is suspected to have immunodeficiency. Irradiation of blood projects is also recommended for in utero transfusions and in patients who have received in utero transfusions (See Chapter 20 Transfusion Practices) [7]. Antifibrinolytics such as aminocaproic acid (Amicar) can be useful if bleeding involves a mucous membrane such as the nose or mouth; Amicar is contraindicated in the setting of urogenital tract bleeding. Specific diagnostic and management strategies will be discussed in the context of the individual diagnoses.
It is beyond the scope of this chapter to describe all of the genes now associated with inherited thrombocytopenia. The discussion that follows will focus on disorders that are likely to have implications for management in infancy, either for treatment of the child or counseling of the family. Disorders will be grouped as classical inherited thrombocytopenia syndromes, congenital syndromes with associated thrombocytopenia, and inherited disorders in which thrombocytopenia is related to abnormalities of vWF and increased platelet destruction. A comparison of diagnostic findings is summarized in Table 15.2.
| Diagnosis | Platelet size | Associated blood cell morphology | Inheritance | Associated Genes | Associated features in childhood |
|---|---|---|---|---|---|
| Wiskott–Aldrich Syndrome | Small | XLR | WASP | Immunodeficiency, eczema | |
| Congenital amegakaryocytic thrombocytopenia | Normal | AR | c-MPL | Reduced or absent megakaryocytes | |
| Thrombocytopenia with absent radii | Normal | Compound heterozygosity | RBM8A | Absent radii | |
| Radioulnar synostosis with amegakaryocytic thrombocytopenia | Normal | AD |
| Radioulnar synostosis | |
| Thrombocytopenia with predisposition to myeloid malignancy | Normal | AD |
| Family history of MDS or leukemia | |
| Bernard–Soulier syndrome | Large | AR | GPIB, GP9 | Platelet dysfunction | |
| MYH9-related disease | Large | WBC inclusions | AD | MYH9 | Family history of renal dysfunction, hearing loss, cataracts |
| Gray platelet syndrome | Large | Platelets with alpha granule deficiency | AR | NBEAL2 | |
| GATA1 mutation | Large | Platelets with alpha granule deficiency | XLR | GATA1 | Anemia |
| Sitosterolemia | Large | Red cell stomatocytes | AR | ABCG5, ABCG8 | Hemolysis, high cholesterol |
| Fanconi anemia | Normal | Red cell macrocytosis | AR/XLR | Multiple | Congenital anomalies, abnormal chromosomal breakage |
| Dyskeratosis congenita | Normal | Red cell macrocytosis | AD/AR/XLR | Multiple | IUGR, cerebellar hypoplasia, immunodeficiency, lacrimal duct stenosis, nail dysplasia, shortened telomeres |
| Paris Trousseau syndrome | Normal | Platelets with fused alpha granules | Sporadic | Del11q23 | Heart defects, developmental delay |
| Noonan syndrome | Normal | AD | PTPN11 | Heart defects, lymphatic malformations | |
| 22q11.2 deletion syndrome | Large | Sporadic | Del 22q11.2 | Heart defects, T-cell dysfunction | |
| von Willebrand disease type 2B | Large | Platelet clumping | AD | VWF | von Willebrand factor deficiency |
| Platelet-type von Willebrand disease | Large | Platelet clumping | AD | GPIB | von Willebrand factor deficiency |
| Thrombotic thrombocytopenic purpura | Large | Schistocytes | AR | ADAMTS13 | Hemolysis, renal insufficiency |
Inherited Thrombocytopenia Syndromes
In these disorders, thrombocytopenia tends to be the presenting finding and mutations directly affect megakaryocyte or platelet production.
Wiskott–Aldrich Syndrome (MIM #301000)
Wiskott-Aldrich syndrome (WAS) is an X-linked disorder in which thrombocytopenia with small platelets is associated with eczema, immunodeficiency and a high risk of EBV-associated lymphoma [8–10]. Wiskott–Aldrich syndrome is rare, with an incidence of approximately one to four cases per 1,000,000 live male births, and an average age at diagnosis of 24 months in the absence of a known family history [11]. The disease is due to mutation in the WAS gene located on chromosome Xp11.23, encoding Wiskott–Aldrich associated protein (WASp), a protein that is important for actin polymerization and cytoskeletal rearrangement [12]. There is considerable variation in the clinical severity in patients with WAS mutations [13]. Thrombocytopenia may be mild or severe, with approximately half of affected boys having platelet counts <20,000/ul; bleeding symptoms generally correlate with the degree of thrombocytopenia and can include gastrointestinal and intracranial hemorrhage [11, 14]. Although eczema is considered part of the classic presentation of WAS, it may not always be present or may develop later in childhood. In one series, fewer than 30% of patients had the complete triad of thrombocytopenia, eczema, and immunodeficiency, and 20% had thrombocytopenia alone [11]. WAS mutations that lead to isolated thrombocytopenia may be referred to as X-linked thrombocytopenia, although mutations in GATA1 also cause thrombocytopenia with X-linked inheritance [15]. In addition to small platelets, laboratory findings that suggest WAS include reduced numbers of T-lymphocytes and abnormal immunoglobulins [16]. The diagnosis can be confirmed by flow cytometry showing reduced or absent expression of WASp in lymphocytes, or mutational analysis of the WAS gene [10]. Management of WAS must take into account not only the bleeding symptoms but also the significant risk for infection, autoimmune manifestations and EBV-associated lymphoma related to the associated immunodeficiency [10, 11]. Patients should be referred for early consideration of hematopoietic stem cell transplant, which is the current standard for definitive therapy. Due to the associated immunodeficiency, transfused blood products including platelets should be irradiated to prevent transfusion-associated graft vs. host disease. Intravenous immunoglobulin therapy is unlikely to improve platelet counts [17], although it is important for prevention of infection and may improve symptoms related to autoimmunity. Splenectomy may improve platelet counts in patients with X-linked thrombocytopenia, although it has been associated with greater risk of bacteremia and death [11]; additionally splenectomy has been associated with an inferior outcome in patients who undergo hematopoietic stem cell transplant [18, 19]. The role of the newer thrombopoietin receptor agonists, such as Eltrombopag and Romiplostim, in patients with Wiskott–Aldrich syndrome remain unclear, though they may be a valuable adjunct as a bridge to transplant [20, 21].
Congenital Amegakaryocytic Thrombocytopenia (MIM #604498)
Congenital amegakaryocytic thrombocytopenia (CAMT) typically presents as thrombocytopenia in an otherwise well infant. CAMT is an autosomal recessive disease caused by mutations in the receptor for thrombopoietin, c-MPL. Thrombopoietin signaling is essential not only for megakaryocyte and platelet production but also for the maintenance of hematopoietic stem cells [22–24]. Fewer than 100 cases of CAMT have been reported in the literature, although more have certainly been diagnosed. At birth, most affected infants have isolated thrombocytopenia. Evaluation of the bone marrow shows a selective deficiency of megakaryocytes, although occasionally initial marrows may appear relatively normal [25]. Over time, most patients will progress to pancytopenia with trilineage marrow aplasia. An increased incidence of MDS and AML is also reported [26]. Genotype–phenotype correlation studies show that patients who have mutations resulting in absent expression of c-MPL (type I mutations) have persistent thrombocytopenia from birth and develop pancytopenia at a median age of 22 months. In comparison, patients whose mutations result in residual expression of c-MPL (type II mutations) may have transient improvement of their platelet counts after birth, followed by a recurrence of thrombocytopenia and a later onset of marrow failure at a median of 28 months [27]. While physical abnormalities have been described in patients with CAMT, there is no specific pattern associated with the diagnosis [27]. Neurologic abnormalities have been seen, including polymicrogyra, underdevelopment of the cerebellum and absence of the corpus callosum [27–29]; some findings might be attributable to sequelae of intracranial hemorrhage [30]. Once suspected, the diagnosis of CAMT is confirmed by genetic testing. If clinically available, a serum thrombopoietin level can be helpful. In the setting of pathogenic c-MPL mutations, serum thrombopoietin levels are very high, often as much as ten-fold above normal, due to absent thrombopoietin uptake and clearance from the circulation [31, 32]. If the clinical picture is consistent with CAMT but thrombopoietin levels are unexpectedly low, then mutations in the gene for thrombopoeitin itself can be investigated [33, 34]. Treatment of CAMT is initially supportive, but hematopoietic stem cell transplant is recommended once there is onset of trilineage marrow failure [27]. In the rare case that CAMT is due to a mutation in thrombopoietin, then treatment with recombinant thrombopoietin or a thrombopoietin receptor agonist may be effective [33].
Thrombocytopenia with Absent Radii (MIM #274000)
Thrombocytopenia with absent radii (TAR) is usually recognized at birth or even prenatally by the eponymous findings of thrombocytopenia and bilateral radial abnormalities [35–37]. The prevalence of TAR is estimated at 1:200,000–1:100,000. Radii may be hypoplastic or absent. Additional limb abnormalities may be seen, but importantly thumbs are present. Affected children may also have congenital heart or urogenital defects. Eosinophilia and cow’s milk allergy are common, and exposure to cow’s milk may exacerbate thrombocytopenia. Defects in platelet function may be present and lead to an increased severity of bleeding symptoms [38, 39]. Leukemoid reactions may occur, leading to concerns about malignant transformation. The differential diagnosis of thrombocytopenia in children with radial hypoplasia or aplasia includes Fanconi anemia. Thumbs are always present in TAR, whereas they may be absent or hypoplastic in Fanconi anemia. Radial aplasia has also been reported in the 22q11 deletion syndrome [40]. The genetic basis of TAR has only recently been elucidated [41, 42]. Affected patients have compound heterozygous mutations affecting the gene RBM8A, which encodes the exon-junction complex subunit member Y14 [43]. Typically, one mutation is a microdeletion on chromosome 1q21.1, encompassing the RBM8A gene, and the second mutation is a noncoding variant involving either the 5’UTR or first intron of the remaining RBM8A allele. Confirmation of the carrier status for the parents can be useful in counseling them regarding recurrence risks should they wish to have more children. Curiously, thrombocytopenia in patients with TAR but usually improves after the first year of life, so most patients can be managed with supportive care, including avoidance of cow’s milk, until platelet counts improve. Data regarding long-term outcomes in patients with TAR are sparse but intermittent thrombocytopenia may recur. Leukemia, both lymphoid and myeloid, has been reported [44, 45].
Thrombocytopenia with Radioulnar Synostosis
Radioulnar synostosis with amegakaryocytic thrombocytopenia (RUSAT), has been attributed to mutations in MECOM (RUSAT2, MIM #616738) as well as HOXA11 (RUSAT1, MIM #605432). MECOM encodes the zinc-finger transcription factor EVI1, which has an important role in hematopoietic stem cell renewal [46–48]. Heterozygous mutations in MECOM have been associated with a range of findings including thrombocytopenia, radioulnar synostosis, clinodactyly, cardiac or renal malformations, B-cell deficiency and hearing loss [49, 50]. Not all patients with MECOM mutations have radioulnar synostosis, and thus some authors suggest that the disease should be called MECOM-associated syndrome, to reflect the variety of clinical presentations [49]. Three patients from two families with thrombocytopenia and radioulnar synostosis were found to have heterozygous mutations in HOXA11 [51]. Treatment of thrombocytopenia is generally supportive, however evolution to trilineage marrow failure or MDS is common [49, 51, 52], and thus identification of potential donors should occur early in the event that a hematopoietic stem cell transplant is needed.
Thrombocytopenia with Increased Risk of Leukemia
Autosomal dominant mutations in several genes are associated with thrombocytopenia with a markedly increased risk for development of myelodysplastic syndrome (MDS) or leukemia. Mutations in RUNX1 (MIM #601399) were the first to be described as causing the syndrome of Thrombocytopenia with Predisposition to Myeloid Malignancy [53], and subsequently mutations in ANKRD26 (MIM #188000) and ETV6 (MIM #616216) were also found to increase the risk for leukemia [54–56]. Thrombocytopenia can be mild and may not be recognized in the newborn period; the family history may lead to initial testing, or low platelets may be discovered incidentally. Patients with RUNX1 mutations may have platelet dysfunction in addition to thrombocytopenia, which can exacerbate bleeding tendencies. The risk of developing MDS or acute myeloid leukemia (AML) may be as high as 50% in affected family members with RUNX1 mutations, and although this most commonly occurs in adults it may occur at any age [53, 57]. Interesting, mutations in RUNX1 are also associated with an increased risk for T-lymphoblastic lymphoma, and ETV6 is associated with MDS, AML, and B-lymphoblastic leukemia [58–60]. Because acquired mutations in RUNX1 and ETV6 are common in MDS and leukemia, it is important to confirm the germline nature of mutations either by identification of the mutation in other family members or in DNA isolated from a non-hematologic tissue such as skin fibroblasts. Management of infants with RUNX1, ANKRD26 or ETV6 mutations includes supportive care for bleeding symptoms and genetic counseling. Older children and adults should have regular monitoring of blood counts and bone marrow studies to identify changes suggesting evolution towards malignancy which would be an indication for hematopoietic stem cell transplant [61]. Family members should receive genetic counseling and mutation testing should be considered regardless of their platelet counts; it is additionally important to clarify the mutational status of any family member being considered as a hematopoietic stem cell donor.
Bernard–Soulier Syndrome (MIM #231200)
Bernard–Soulier syndrome (BSS) is caused by mutations that affect the expression of the GPIB/IX/V complex on the platelet surface, which serves as the receptor for von Willebrand factor (vWF) [62, 63]. It is inherited as an autosomal recessive disorder, although rare cases of monozygous mutations have been described [64, 65]. The prevalence of BSS is estimated at 1:1,000,000. Patients with BSS have large platelets and bleeding symptoms that are out of proportion to the degree of thrombocytopenia, due to the impaired binding of platelets to vWF. It is important to recognize the platelet dysfunction in patients with BSS and not use the platelet count alone to make clinical decisions regarding hemostasis. Management is supportive; however, while platelet transfusions are effective, they should be reserved for clinically significant bleeding or major procedures due to the risk for alloimmunization, with development of anti-HLA or -GPIb/IX antibodies that might limit efficacy of future transfusions [66]. To minimize the risk for alloimmunization, transfused platelets should be leukocyte reduced and, if feasible, HLA matched. Antifibrinolytics can be particularly helpful as platelet-sparing agents in management of mucous membrane bleeding. Desmopressin or DDAVP, which may improve hemostasis in patients with patients with platelet function abnormalities, should not be used in infants due to the risk of hyponatremia. The role of rFVIIa is unclear, however there are reports supporting its efficacy in patients with BSS where platelets are not effective [67–69].
MYH9-Related Disease (MIM #155100)
Macrothrombocytopenia that is inherited in an autosomal dominant fashion is seen in MYH9-related disease [70]. The prevalence is unknown, though it is one of the more common causes of familial thrombocytopenia in the Italian registry [5]. The identification of mutations in MYH9, a non-muscle myosin heavy chain gene, allowed reclassification of several diseases, including May–Hegglin anomaly, Sebastian platelet syndrome, Fechtner syndrome, Epstein syndrome, and autosomal deafness DFNA17, as variants of a single disorder [71, 72]. In addition to large and frequently giant platelets, which may be larger than erythrocytes, careful evaluation of the peripheral smear may reveal Dohle-like white cell inclusions. Where available, detection of these inclusions is enhanced by the use of immunofluorescent staining techniques [4]. Genetic testing may not be necessary to make the diagnosis. Treatment of thrombocytopenia is supportive, however, the majority of the morbidity in MYH9-related disease comes not from bleeding symptoms but from the late onset of renal disease, sensorineural deafness, or cataracts. Renal disease, characterized by progressive proteinuric nephropathy, develops in approximately 25% of patients and may progress to renal failure [73]. Although these complications are not anticipated during infancy, families should be educated about the importance of ongoing follow up. Parents should be referred for evaluation if they have not already been tested.
Gray Platelet Syndrome
Gray platelet syndrome can be suspected on the basis of laboratory findings, including thrombocytopenia with large platelets that are pale in appearance on a peripheral blood smear. The pale appearance of the platelets is due to lack of intracellular alpha granules, which contain a diverse array of compounds released on platelet activation such as vWF, thrombospondin, factor V, fibroblast growth factor and others [74]. The absence of platelet alpha granules can be confirmed by electron microscopy. Two different inheritance patterns have been associated with gray platelet syndrome. Mutations in NBEAL2 (MIM #139090), encoding a protein important for intracellular vesicle trafficking, are inherited in an autosomal recessive fashion [75–77]. Mutations in the hematopoietic transcription factor GATA1 (MIM #300367, #314050) are inherited as X-linked recessive traits [78]. GATA1 regulates the expression of NBEAL2 [79], thus explaining the overlapping phenotypes resulting from mutations in these different genes. As GATA1 regulates genes critical for both megakaryopoiesis and erythropoiesis, it is not surprising that mutations in this transcription factor may also be associated with a variety of red cell abnormalities, including dyserythropoeisis, thalassemia, or even porphyria [15]. Because many of the components of the alpha granules have roles in hemostasis, platelet function in gray platelet syndrome is abnormal [80]. Bone marrow biopsy would not typically be pursued in the newborn period; however, marrows performed in older patients may show myelofibrosis and megakaryocyte emperipolesis [80, 81]. Management involves supportive care.
Congenital Syndromes with Associated Thrombocytopenia
In many of the inherited bone marrow failure syndromes, cytopenias are not typically present at birth but the diagnosis may be suspected on the basis of other congenital abnormalities or a family history [82]. Markers of stress erythropoiesis, such as elevated fetal hemoglobin or macrocytosis, which can be useful screening tools in older patients, are less helpful in newborns. In other congenital syndromes, not classified as marrow failure disorders, thrombocytopenia may be an associated finding.
Fanconi Anemia (MIM 607139)
Fanconi anemia is an inherited bone marrow failure syndrome caused by a defect in DNA repair (reviewed in [83]). In addition to cytopenias, patients with Fanconi anemia commonly have congenital abnormalities and are at a significantly increased risk for cancer. The prevalence of Fanconi anemia is estimated at 1–5 per million, and is more common in people of Ashekanzi Jewish descent, the Roma population of Spain and black South Africans due to the presence of founder mutations in those populations [84–86]. Bone marrow failure most commonly develops in the first decade of life, with a median age of 7 years, but occasionally may be seen in the newborn period. Thrombocytopenia may be the presenting hematologic abnormality [82]. More frequently, infants with Fanconi anemia are recognized on the basis of associated congenital anomalies such as poor growth, thumb or forearm abnormalities, café au lait spots, VACTERL syndrome (Vertebral defects, Anal atresia, Cardiac defects, Tracho-Esophageal fistula, Renal anomalies, and Limb abnormalities) and others. Fanconi anemia is diagnosed by demonstration of abnormal chromosomal breakage in the presence of clastogens such as diepoxybutane or mitomycin C. Mutations involving more than 18 genes have been identified [83], definining subypes of the disease. Eighty to 90% of cases are due to mutations in FANCA, FANCC, or FANCG. Inheritance is autosomal recessive, with the exception of FANCB which is inherited in an X-linked recessive pattern [87]. Although the diagnosis is made by demonstration of chromosomal fragility, confirmation of the subtype is important for counseling. Due to the increased risk for cancers, including hematologic and non-hematologic malignancies, patients with Fanconi anemia require ongoing follow up and screening to maximize opportunities for early detection when outcomes are best [83, 88].
Dyskeratosis Congenita
Dyskeratosis congenita is an inherited marrow failure syndrome caused by defects in telomere maintenance. The classic clinical triad consists of skin and nail abnormalities, oral leukoplakia and bone marrow failure, but in fact this triad is present in only a minority of patients; other symptoms such as pulmonary or liver fibrosis, strictures or premature graying may also be seen, and patients are at increased risk of developing malignancy [89, 90]. Most commonly, symptoms in dyskeratosis congenita do not present until later in life, but severe mutations may present in infancy. Hoyeraal–Hreidarsson syndrome is a severe form of dyskeratosis congenita diagnosed in infancy with findings including cytopenias, intrauterine growth retardation, cerebellar hypoplasia, developmental delay, immunodeficiency, and lacrimal duct stenosis [91–94]. Another severe variant of dyskeratosis congenita that can present in early childhood is Revesz syndrome, characterized by exudative retinopathy and intracranial calcification in addition to the features above, and associated with mutations in TINF2 [89, 95]. The diagnosis of dyskeratosis congenita can be confirmed by the finding of very short telomeres. Mutations in at least 11 genes associated with telomere maintenance are recognized, including DKC1 (MIM #305000), TERT (MIM #613989), and others, though not all causative mutations have been identified [90, 96]. Inheritance varies and can include autosomal dominant, autosomal recessive and X-linked recessive patterns. Children with Hoyeraal–Hreidarsson syndrome are at risk for death in early childhood due to the complications of marrow failure unless they can be successfully transplanted [97].
Paris Trousseau Syndrome (MIM #188025)
Paris Trousseau syndrome (PTS) is a variant of Jacobsen syndrome (MIM 147791) in which thrombocytopenia accompanies congenital cardiac defects such as hypoplastic left heart syndrome and developmental delay [98–101]. Cytogenetic studies show deletion of the terminal portion of chromosome 11q23, encompassing the locus for the megakaryocytic transcription factor FLI1 which is responsible for the platelet phenotype [100, 102, 103]. Bone marrow findings may include micromegakaryocytes, and ultrastructural studies of platelets may show giant, fused alpha granules and a reduction of dense granules [104]. Consistent with the importance of FLI1 in PTS, isolated mutations in FLI1 have been identified in families with thrombocytopenia and platelet granule abnormalities resembling those seen in patients with PTS [105, 106]. Treatment is generally supportive. Thrombocytopenia tends to improve later in infancy, though platelet function may remain abnormal [107].
Noonan Syndrome
Noonan syndrome is an autosomal dominant disorder associated with abnormal facies, developmental delay, short stature, congenital heart defects, renal anomalies, lymphatic malformations, and bleeding disorders [108]. Its estimated prevalence is between 1:1,000 and 1:2,500 living births. Noonan syndrome is caused by a gain of function mutation in PTPN11, encoding a cytoplasmic protein tyrosine phosphatase called SHP2 that has important regulatory roles in the Ras/MAPK and PI3K pathways [109]. Thrombocytopenia is not usually the dominant symptom in Noonan syndrome, but infants may present with very low platelets [110, 111]. In addition to thrombocytopenia, laboratory abnormalities of hemostasis, including reduced coagulation and platelet function, have been described in the majority of patients [112]. Children with Noonan syndrome are at increased risk for malignancy, such as juvenile myelomonocytic leukemia (JMML) [113, 114] and other cancers [114].
The 22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome is one of the most commonly diagnosed chromosomal microdeletion syndromes in humans and is associated with a variety of abnormalities including facial abnormalities, palatal abnormalities, cardiac defects and thymic hypoplasia or aplasia [115]. Mild macrothrombocytopenia is commonly associated with the 22q11.2 deletion syndrome, and is thought to be related to heterozygous deletion of the GPIBB gene, located within the deleted portion of chromosome 22 [116, 117]. In the majority of patients, this thrombocytopenia does not result in clinically significant bleeding. Rarely, co-inheritance of a second mutation in GPIBB can lead to true Bernard–Soulier syndrome [118]. Patients with 22q11.2 deletion syndrome are also at increased risk of immune thrombocytopenia (ITP), given their underlying T-cell dysfunction [119], so if thrombocytopenia follows an acquired course, ITP should be considered.
In addition to the congenital disorders described above, thrombocytopenia is common in infants with trisomies 13, 18, and 21. In the first week of life, 83% of infants with trisomy 18 and 75% of those with trisomy 13 were noted to be thrombocytopenic [120]. Several metabolic disorders are associated with thrombocytopenia. Lysosomal storage diseases such as Gaucher disease may present in the newborn period with low platelets [121, 122], and thrombocytopenia may be part of the clinical spectrum in mitochondrial disorders [123]. Sitosterolemia, or Mediterranean macrothrombocytopenia, is caused by mutations in ABCG5 or ABCG8, which encode the sterol efflux transporters sterolin-1 and -2, resulting in accumulation of phytosterols. Affected patients have macrothrombocytopenia and hemolytic anemia, and a review of the smear may show stomatocytes [124–127]. Symptoms may develop in infancy, potentially exacerbated by the use of soy-based formulas or intralipids [128]. While rare, recognition and treatment of an underlying metabolic disorder can avoid important extrahematopoietic complications and may mitigate thrombocytopenia.
Inherited Disorders Associated with Abnormalities of vWF
In some cases, familial thrombocytopenia is associated with increased platelet consumption due to abnormalities of vWF. Examples include type 2B von Willebrand disease (vWD; MIM #613554) and congenital thrombotic thrombocytopenic purpura (TTP; MIM #274150). Thrombocytopenia in these disorders is variable but may be exacerbated in the newborn period given that expression of vWF is increased at birth due to the effects of maternal estrogens, and women may have worsening of their thrombocytopenia during pregnancy for the same reason. Type 2B von Willebrand disease is caused by an activating mutation of VWF that increases its affinity for GPIB on platelets, resulting in increased platelet clearance and loss of high molecular weight vWF multimers. A reciprocal mutation in GPIB that increases its affinity for vWF has a similar phenotype and is called platelet-type vWD. In both disorders, platelet clumps that represent large complexes between platelets and the abnormal vWF may be visible on the peripheral blood smear [129]. The inheritance pattern is autosomal dominant. Bleeding is related primarily to the deficiency of vWF but in severe cases thrombocytopenia may contribute [130]. The diagnosis is suspected if the patient has both low vWF activity and thrombocytopenia; additional laboratory findings may include the loss of high molecular weight vWF multimers. Genetic studies can distinguish between type 2B vWD, and platelet-type vWD. This is most readily achieved by sequencing of vWF exon 28 for the pathogenic variant vWF c.3922C>T, p.Arg1308Cys [130]. Bleeding in patients with type 2B vWD is managed with vWF-containing products and antifibrinolytics [129, 130]. DDAVP is not appropriate in infants due to the risk of hyponatremia and it can exacerbate thrombocytopenia in patients with type 2B disease. In platelet-type vWD, vWF replacement is not effective and platelet transfusion should be used instead [131].
In congenital TTP, deficiency of the von Willebrand cleavage factor ADAMTS13 results in the formation of ultralarge von Willebrand factor multimers and a non-immune, microangiopathic hemolytic anemia. The finding of schistocytes on the peripheral blood smear is the hallmark of this diagnosis. The associated hemolytic anemia may lead to prolonged neonatal jaundice, and renal insufficiency or neuropsychiatric symptoms may also be present [132, 133]. Beyond the newborn period, symptoms may be episodic. Inheritance is autosomal recessive and the diagnosis can be confirmed by the finding of severe deficiency of ADAMTS13 (activity <10%) and identification of homozygous or compound heterozygous mutations. Treatment of congenital TTP includes plasma infusion, to replace the missing ADAMTS13 enzyme activity. Plasma infusions are given daily until clinical stabilization and then prophylactically to control symptoms [134]. The platelet count can be used as a marker of clinical response. Novel approaches to ADAMTS13 replacement, including recombinant products and gene therapy, are in development [135].
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


