Abstract
The ‘classical’ Philadelphia chromosome-negative myeloproliferative neoplasms (MPN), essential thrombocythaemia (ET), primary myelofibrosis (PMF) and polycythaemia vera (PV), are characterized by clonal myeloproliferation with effective maturation causing accumulation of terminally differentiated cells in the peripheral blood and/or splenomegaly. Although each disease has distinct clinical manifestations clonal haematopoiesis is driven, in most cases, by upregulation of the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway [1]. Polycythaemia vera and ET are relatively indolent, with most patients having nearly normal life expectancy, in contrast to PMF [2]. Thrombosis and haemorrhage are the main causes of morbidity and mortality in both PV and ET, and evolution to myelofibrosis (MF) and/or accelerated/blast phase (AP/BP) is estimated to occur in 10% of patients [3, 4]. Overt (classical) PMF is the most aggressive of the three diseases, with a median overall survival of five years. The most common causes of mortality are transformation to BP (20 to 25% of patients), thrombosis, cardiovascular complications and infections [5]. However, it is important to note that incidence figures, survival and also risk-factor determinations may be inaccurate and at times conflicting because of the inadvertent labelling in some studies of patients with prefibrotic/early PMF (prePMF) or ‘masked’(prodromal) PV as ET [6–8].
Introduction
The ‘classical’ Philadelphia chromosome-negative myeloproliferative neoplasms (MPN), essential thrombocythaemia (ET), primary myelofibrosis (PMF) and polycythaemia vera (PV), are characterized by clonal myeloproliferation with effective maturation causing accumulation of terminally differentiated cells in the peripheral blood and/or splenomegaly. Although each disease has distinct clinical manifestations clonal haematopoiesis is driven, in most cases, by upregulation of the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway [1]. Polycythaemia vera and ET are relatively indolent, with most patients having nearly normal life expectancy, in contrast to PMF [2]. Thrombosis and haemorrhage are the main causes of morbidity and mortality in both PV and ET, and evolution to myelofibrosis (MF) and/or accelerated/blast phase (AP/BP) is estimated to occur in 10% of patients [3, 4]. Overt (classical) PMF is the most aggressive of the three diseases, with a median overall survival of five years. The most common causes of mortality are transformation to BP (20 to 25% of patients), thrombosis, cardiovascular complications and infections [5]. However, it is important to note that incidence figures, survival and also risk-factor determinations may be inaccurate and at times conflicting because of the inadvertent labelling in some studies of patients with prefibrotic/early PMF (prePMF) or ‘masked’(prodromal) PV as ET [6–8].
The diagnostic approach to classifying MPN subtypes incorporates clinical features, peripheral blood counts and smear analysis, bone marrow (BM) morphology, karyotype and molecular genetic tests [2]. Following this concept, the detection of one of the characteristic driver mutations such as JAK2V617F, JAK2 exon 12, MPL and calreticulin (CALR) is a major diagnostic feature. JAK2 mutations are detected in more than 90% of patients with PV and are therefore used as a highly sensitive clonal marker in this subtype. However, JAK2 mutations may also occur in ET and PMF, while CALR is virtually not seen in PV [9]. Therefore BM morphology remains the central diagnostic platform and is essential for distinguishing ET from prePMF and diagnosing cases that do not express JAK2, MPL or CALR (so called ‘wild-type’ or ‘triple-negative’ MPN). It is essential to emphasize that, because not a single histological parameter is restricted to a given subgroup, morphological BM patterns, although very important, should only be interpreted in the light of appropriate clinical, laboratory and molecular findings [6, 8, 10, 11]. Along these lines, standardized assessment of the megakaryocytic histotopography, i.e. their arrangement within the BM space, and megakaryocytic morphology such as the identification of certain nuclear abnormalities and maturation defects, remain crucial to the morphological diagnosis of MPN [12, 13]. In terms of routine clinical practice, mutation screening in ET or PMF should start with JAK2V617F, followed by CALR mutations in JAK2-unmutated patients, whereas MPL mutation screening is best reserved for JAK2 and CALR unmutated cases. In PV mutation screening should start with JAK2V617F because it represents more than 95% of all JAK2 mutations. Other JAK2 mutations, including JAK2 exon 12 mutations, are recommended only in the absence of JAK2V617F and presence of a subnormal serum erythropoietin level.
Initial Stages of MPN
The diagnosis of initial MPN may be clinically challenging, particularly because typical haematological features and symptoms generally found in overt stages of MPN are often not present in the early stages. Overlapping features, e.g. thrombocytosis, can be seen in all three diseases and compound the issue [8]. However, careful assessment of BM features and integration of molecular data (JAK2, CALR and MPL) help in guiding the differential diagnosis in the early disease stage [11]. A precise disease diagnosis has clinical relevance. Although evolution to overt myelofibrosis and blastic transformation might also occur in ET patients, the overall risk is significantly lower than in prePMF patients. Furthermore, identification of initial stage MPN enables early therapeutic intervention to prevent disease-related complications. It is important to recognize that major thrombotic and other complications can sometime precede a definite clinical diagnosis of MPN. Along these lines, patients with positive driver mutation status and cerebral or splanchnic vein thrombosis including Budd–Chiari syndrome but without clinical features of overt MPN have been described [14].
Essential Thrombocythaemia
Epidemiology
Essential thrombocythaemia is characterized by a sustained thrombocytosis that is not associated with any of the known reactive causes. The age distribution is broad, ranging from adolescent to old patients; most cases are seen in patients older than 50 years [4], but well-documented cases in young adults and children have also been reported. Rare familial forms of ET may also occur. While patients with ET have a near normal life expectancy, they are at increased risk of thrombosis or haemorrhage. Nearly 90% of patients with ET have a mutation in one of three genes: 50 to 60% have a JAK2V617F mutation, 25 to 30% a mutation in CALR and 3 to 5% in MPL. Mutations in all three genes lead to aberrant activation of the JAK-STAT pathway. Additional mutations and cytogenetic abnormalities are uncommon in ET (<10% of patients) and have not been shown to be correlated with worse prognosis.
Clinical Features
Many patients are asymptomatic and are diagnosed incidentally after a routine blood count shows an elevated platelet count. However, a thrombotic or haemorrhagic event is often the first sign of ET. Symptomatic patients can experience headache and paresthesia, dizziness, lightheadedness, acroparesthesia, livedo reticularis or erythromelalgia. Some patients also present with mild splenomegaly, elevated white blood cell counts or, in some cases, mild anaemia; however, the presence of these findings suggests a need for a careful differentiation from prePMF [6, 8, 15]. The thrombocytosis in ET has been associated with potential thrombotic and bleeding complications. Both arterial and venous thrombosis can occur, with the former being more common. The venous clots commonly involve unusual sites particularly in the splanchnic beds such as the portal or mesenteric vein [16, 17].
Morphology
Peripheral Blood
In ET peripheral blood smears reveal an overall excess of platelets that often display anisocytosis, ranging from tiny forms to abnormally large or giant platelets that may reveal bizarre shapes, pseudopods and agranularity. The mean platelet volume is markedly increased due to the large platelet size, while the observed high platelet distribution width reflects the wide spectrum of platelet sizes. Circulating megakaryocyte nuclei and cytoplasmic fragments may be observed in cases of ET. The white blood cell count and leukocyte differential are usually normal. Following iron deficiency due to recurrent haemorrhage, the red cells may be hypochromic and microcytic [13, 18].
Bone Marrow
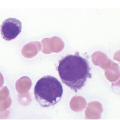
Characteristic BM histopathology in ET includes an age-matched normal marrow cellularity, while moderate hypercellularity is only seen in a small fraction of patients (Figure 10.1) that is accompanied by increased megakaryopoiesis with a predominance of mature forms, without significant erythroid or granulocytic myeloproliferation [6, 13, 15, 19]. Gross disturbances of the topographic distribution of the megakaryocytes, such as their arrangement to form dense clusters or a prominent lining along the bony trabeculae, are normally not detectable. In ET megakaryocytes show a random distribution or very loose groupings within the BM space. Megakaryocyte cytology includes mostly large to giant forms, displaying abundant mature cytoplasm and deeply lobulated and hyperlobulated (staghorn-like) nuclei. Emperipolesis of neutrophils is frequently noted within the megakaryocyte cytoplasm. Bizarre, highly atypical megakaryocytes or frequent dense clusters, such as those observed in prePMF (Figure 10.2), are not found in ET and, if present, the diagnosis of ET should be questioned [19]. The myeloid to erythroid ratio is close to normal; however, in a few cases a proliferation of erythroid precursors may be found, particularly if the patient has experienced recurrent major bleedings, but granulocytic proliferation is highly unusual [13]. The network of reticulin fibres is usually normal or very rarely (less than 5%) minimally (grade 1) increased in ET, and the finding of significant reticulin fibrosis or any collagen fibrosis excludes the diagnosis of ET. The morphological findings in the BM biopsy are essential to distinguish ET from other MPN subtypes or myeloid disorders and reactive conditions that present with sustained thrombocytosis. The finding of even a mild degree of combined granulocytic and erythroid proliferation should raise consideration of the prodromal stage of PV, and the finding of granulocytic proliferation associated with bizarre, highly atypical megakaryocytes and dense clustering should prompt concern for prePMF and should be discriminated by careful examination, taking into consideration the so-called minor clinical findings [8].
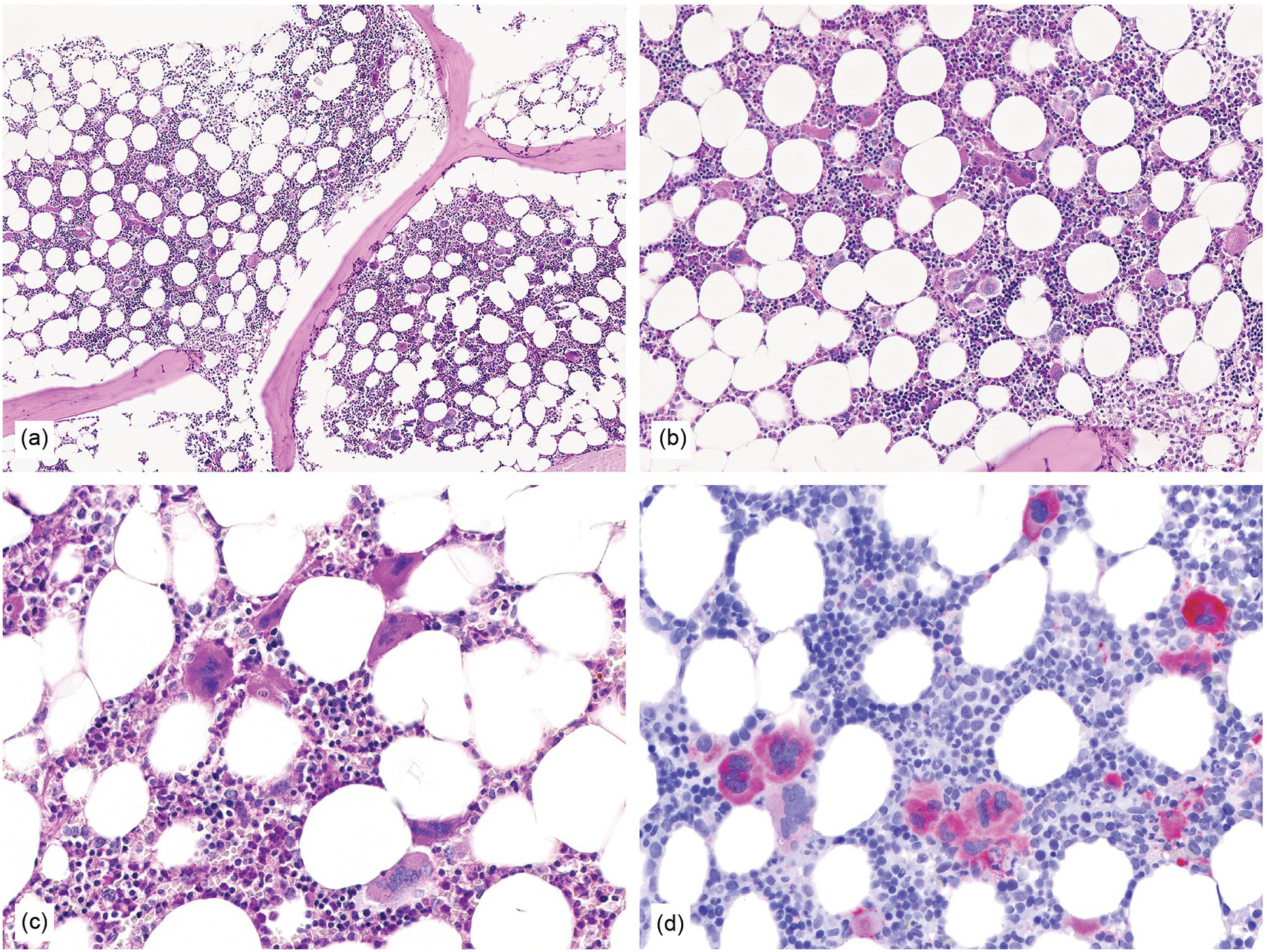
Figure 10.1 (a–d) Bone marrow in essential thrombocythaemia (ET): age-matched cellularity, except for a prominent increase in large to giant megakaryocytes loosely clustered or dispersed throughout the bone marrow space. No proliferation and/or significant left shift of neutrophil granulopoiesis or erythropoiesis surrounding giant mature megakaryocytes is present. Note the conspicuously increased number of large megakaryocytes without maturation defects showing hyperlobulated, occasionally staghorn-like nuclei. No increase in reticulin fibres, but giant megakaryocytes with deep folding of their nuclei.
(a, b): HE; (c): PAS; (d): CD61.
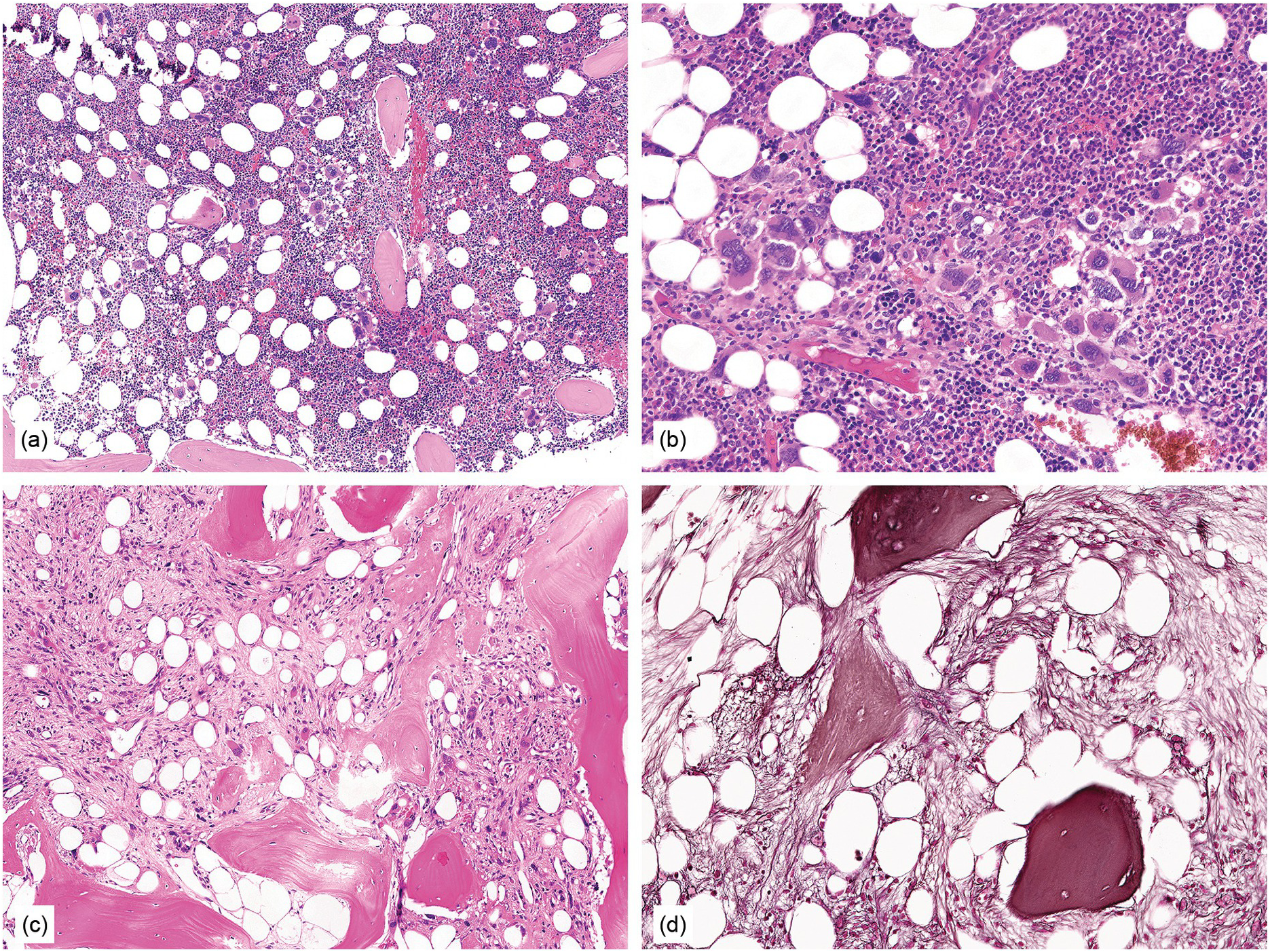
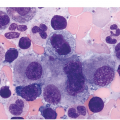
Figure 10.2 Bone marrow in prefibrotic/early primary myelofibrosis and overt primary myelofibrosis. (a, b) Prefibrotic/early primary myelofibrosis: remarkable hypercellurarity revealing large clusters of megakaryocytes abnormally dislocated towards the trabecular bone. In addition to megakaryocyte proliferation there is a conspicuous increase in neutrophil granulopoiesis accompanied by reduction of nucleated erythroid precursors. Clustered small to giant megakaryocytes show a prevalence of striking maturation defects including cloud-like, hypolobulated and hyperchromatic nuclei with only some irregular folding. No significant increase in reticulin fibres. (c, d) Overt primary myelofibrosis (PMF): the marrow haematopoietic cells show a streaming-like pattern of distribution due to fibrosis. In addition, the marrow reveals atypical megakaryocytes, initial osteosclerosis and only a very few erythrocyte precursors. (d) Overt myelofibrosis with bundles of collagen and a dense network of reticulin associated with osteosclerosis.
The exact incidence of myelofibrotic transformation in ET persists as a controversial issue and is certainly influenced by risk status and applied diagnostic criteria, but is in general very low [20]. Careful BM evaluation is crucial in identifying progression to post ET myelofibrosis (post-ET MF), since the appearance of BM fibrosis with a grade MF-2 or MF-3 is a prerequisite for the diagnosis.
Differential Diagnosis and Work-Up
Because the clinical presentation and symptoms of ET are similar to those of other MPN, the diagnosis can be challenging. The WHO criteria [2] include sustained platelet counts ≥450 × 109/L and a BM biopsy showing predominantly large to giant, mature megakaryocytes (Table 10.1). Exclusion of reactive causes of thrombocytosis as well as another MPN, such as prePMF and of MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T) is also required, particularly when myelodysplastic features are present. The differential diagnosis includes also cases of early stage PV with associated thrombocytosis that might mimic ET. In these cases, the marrow cellularity and its composition and the cytologic characteristics of the megakaryocytes usually allow separation from ET [6, 19]. Red cell mass studies, erythropoietin level determination and/or endogenous erythroid colony formation analysis may also be indicated in such instances. Testing for JAK2V617F, CALR and MPL mutations is mandatory to rule out reactive thrombocytosis. In rare ‘triple negative’ cases testing for additional myeloid neoplasms associated mutations may also be considered.
| Essential thrombocythaemia |
|---|
Major criteria
Minor criteria
|
| Diagnosis of ET requires meeting all four major criteria or the first three major criteria and one of the minor criteria |
Prognosis and Predictive Factors
Thrombosis and haemorrhage are the major risk factors for ET and the most common cause of death in these patients [16]. Well-established risk factors for major thrombosis include age older than 60 years, cardiovascular risk factors (diabetes, hypertension and smoking), previous thrombosis and the presence of JAK2V617F mutation [16, 21]. Platelet count has not been shown to correlate with thrombotic risk, but platelet counts >1,500 × 109/L may suggest acquired vWF deficiency and an increased risk of bleeding.
At times, the clinical and morphological differentiation between ET and prePMF with associated thrombocytosis might not be clear cut [6]. As the therapeutic relevance in these early stages is still a matter of debate, a strict adherence to the WHO guidelines for making a working diagnosis and close monitoring of the patient to capture any substantial changes that might warrant revision of diagnosis is recommended. In cases presenting simultaneously with myelodysplastic and myeloproliferative features in the BM and more than 15% ring sideroblasts in the aspirate, the diagnosis of MDS/MPN-RS-T should be rendered [2]. It is important to emphasize that the diagnostic criteria for MDS/MPN-RS-T include not only an elevated platelet count in conjunction with anaemia and the presence of ring sideroblasts in the marrow, but also morphologically abnormal megakaryocytes. In a significant proportion of these cases, the presence of co-mutation of SF3B1 and JAK2V617F can be observed.
Primary Myelofibrosis
Epidemiology
Myelofibrosis (MF) can occur either as a primary (PMF) disease or as a secondary disease in the course of PV (post-PV MF) or (in very few cases) of ET (post-ET MF) [3]. Primary myelofibrosis is a clonal myeloid cell proliferation characterized in the fully developed stage by a leukoerythroblastic blood picture, splenomegaly associated with extramedullary haematopoiesis (EMH), myelofibrosis and a megakaryocyte proliferation with atypia in the BM. Primary myelofibrosis eventually leads to cytopaenias and disease-related constitutional symptoms. It usually affects middle-aged to elderly individuals and is only rarely seen in younger people. Primary myelofibrosis is very rare in children. There is an approximately equal incidence in males and females. Mutations in JAK2, MPL, or CALR are present in 90 to 95% of patients with PMF [9]. Recent evidence suggests that each mutation is associated with slightly different clinical manifestations. Patients with CALR-positive PMF are typically younger, have lower leukocyte counts and higher platelet counts compared with JAK2V617F-positive PMF [22, 23]. In addition to the characteristic driver mutations, other co-mutations with detrimental prognostic impact have been identified in a proportion of cases [24].
Clinical Features
Patients present with a spectrum of signs and symptoms, ranging from few to no symptoms in the early stages to severe and debilitating symptoms in the later stages. Constitutional symptoms including fatigue, weight loss, low-grade fever, night sweats and pruritus are common. Extramedullary haematopoiesis inevitably manifests as splenomegaly (in 80% of patients) and hepatomegaly (40 to 70% of patients) but can also cause other problems including pulmonary hypertension, ascites, pericardial tamponade, portal hypertension or splenic infarcts. A peripheral blood smear will be remarkable for the presence of leukoerythroblastosis, i.e. the presence of immature myeloid cells and circulating nucleated red blood cells usually associated with teardrop-shaped erythrocytes (dacrocytes). Patients often present with severe anaemia, and sometimes leukocytosis or thrombocytosis; although in the later stages thrombocytopaenia and leukopaenia are more common [25]. Other findings include elevated serum lactate dehydrogenase (LDH), elevated vitamin B12-binding proteins and hyperuricaemia.
Morphology
Peripheral Blood
In PMF a distinction should be made between the different stages of disease evolution [13, 26]. While prePMF may only very rarely show a single circulating erythroblast or myeloid precursor in a peripheral blood smear characterized by the presence of thrombocytosis, fibrotic PMF is characterized by leukoerythroblastosis. A small but variable number of circulating myeloblasts is observed. In most cases, these cells represent <5% of the WBC; however, a blast count of up to 9% (in the absence of accelerated/blastic transformation) may be observed. The clinical significance of this findings will be discussed later in the chapter. Megathrombocytes (or giant platelets, defined as larger than a red blood cell) and bare megakaryocytic nuclei are frequently observed. Teardrop erythrocytes have been noted to decrease in number following splenectomy or institution of chemotherapy. Anaemia is frequent and, in approximately 60% of patients, the haemoglobin level drops below 10 g/dL. Leukopaenia is seen in about 25% of patients, while leukocytosis is seen in one third. In overt cases thrombocytopaenia may be observed whereas marked thrombocytosis is seen rarely in late-stage disease. A small subset of patients with PMF can present at onset or develop during their disease a sustained absolute monocytosis in peripheral blood associated with morphologic changes in marrow resembling those typically seen in patients with chronic myelomonocytic leukaemia. This occurrence, which is associated with poor prognosis, is generally considered to be evidence of advanced stage and disease progression [11, 27, 28].
Bone Marrow
The histopathology of BM biopsy samples in prePMF usually reveals age-adjusted hypercellularity characterized by a prominent neutrophilic, granulocytic and megakaryocytic proliferation usually associated with a slight reduction of erythropoiesis without dysplastic features, in the absence of or only with minor BM fibrosis [8]. Therefore the demonstration of reticulin fibrosis is not a required criterion for the diagnosis of prePMF [13]. However, this early phase is often undocumented and by the time of the initial biopsy most patients show already a more advanced picture characterized by patchiness of the haematopoietic cellularity with alternately cellular (non-fibrotic) and less cellular, fibrotic areas [13].
The megakaryocytes in prePMF and overt PMF are characterized by the presence of cytological atypia to a more pronounced degree than that seen in any other MPN, particularly ET [13, 19]. The identification of abnormal megakaryocytic histotopography (distribution within the BM space) and atypia is key to the recognition of prePMF. The megakaryocytes often form dense clusters of variable size that are frequently adjacent to BM vascular sinuses and bony trabeculae. Most megakaryocytes are enlarged, but small megakaryocytes may also be seen. Deviations from the normal nuclear:cytoplasmic ratio (an expression of defective maturation), abnormal patterns of chromatin clumping with bulbous, ‘cloud-like’, ‘clumsy’ or ‘balloon-shaped’, hyperchromatic nuclei, and the frequent occurrence of bare (naked) megakaryocytic nuclei are all typical features (Figure 10.2). The megakaryocytes are more often surrounded by granulocytic cells than by erythroblasts. The majority of cases with prePMF eventually progress to overt fibrotic/sclerotic myelofibrosis associated with evidence of EMH [29].
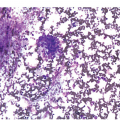
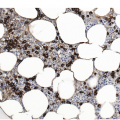
In advanced stages, the BM biopsy shows overt reticulin or collagen fibrosis (fibrosis grades 2 and 3) often associated with different degrees of osteosclerosis (Table 10.2; Figure 10.3). Development of marrow fibrosis is a secondary response to the clonal proliferation of haematopoietic stem cells. Collagen type 3, also known as reticulin, and collagen type 1 are the predominant extracellular components of marrow fibrosis in PMF. These matrix components are produced by marrow fibroblasts not belonging to the malignant clone. The deposition of collagen is a result of the release of fibrogenic cytokines by abnormal megakaryocytes and monocytes derived from the malignant stem cell population. However, fibrosis of the marrow is not unique to PMF and may be secondary to many other disorders [30]. The BM may still be focally hypercellular, but more often is normocellular or hypocellular, with patches of active haematopoiesis alternating with hypocellular areas of loose connective tissue and/or fatty marrow. Atypical megakaryocytes are always present; these occur in large clusters or sheets, often within dilated vascular sinuses together with immature haematopoietic cells. Distended marrow sinusoids containing intravascular haematopoietic cells and megakaryocytes are characteristic findings observed in advanced stage PMF [31]. Occasionally the BM is almost devoid of haematopoietic cells, showing mainly dense reticulin and collagen fibrosis, with small islands of nucleated haematopoietic precursors situated mostly within vascular sinuses. Osteoid seams or appositional new bone formation in bud-like endophytic plaques may be observed. In this osteosclerotic phase, the bone may form broad, irregular trabeculae that can occupy >50% of the BM space, consistent with late-stage disease [29]. Lymphoid aggregates predominantly composed of reactive T-lymphocytes are also seen in many patients.
| Grade | BM fibrosis | BM collagen deposition | BM osteosclerosis |
|---|---|---|---|
| 0 | Scattered linear reticulin with no intersections (cross-overs) corresponding to normal bone marrow | Perivascular collagen only (normal) | Regular bone trabeculae (distinct paratrabecular borders) |
| 1 | Loose network of reticulin with many intersections, especially in perivascular areas | Focal paratrabecular and/or central collagen deposition without connecting meshwork | Focal budding, hooks, spikes or paratrabecular apposition of new bone |
| 2 | Diffuse and dense increase in reticulin with extensive intersections, occasionally with focal bundles of thick fibres mostly consistent with collagen,a and/or focal osteosclerosisb | Paratrabecular and/or central deposition of collagen with focally connecting meshwork or generalized paratrabecular apposition of collagen | Diffuse paratrabecular new bone formation with thickening of trabeculae, occasionally with focal interconnections |
| 3 | Diffuse and dense increase in reticulin with extensive intersections and coarse bundles of thick fibres mostly consistent with collagena, usually associated with osteosclerosisb | Diffuse (complete) connecting meshwork of collagen | Extensive interconnecting meshwork of new bone with overall effacement of marrow spaces |
a If the pattern is heterogeneous, the final score is determined by the highest grade present in at least 30% of the marrow area.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


