Chapter 65 Central Nervous System Tumors in Children
This is a pivotal time for RT in the treatment of children with brain tumors. There are fundamental questions that need to be addressed, including the indications for RT in the setting of clinical and molecular risk stratification for children with medulloblastoma and potentially ependymoma and the refinement of total dose and treatment volumes regimens for specific patient groups, including very young children and those with germ cell tumors. Improved knowledge of treatment-related side effects and a wider availability of proton therapy will likely impact how RT is administered and perceptions about the safety of RT in these patients. Despite the increasing availability of advanced treatment technology, children should be treated on clinical trials designed to improve tumor control, objectively study CNS effects, and build a consensus for treatment guidelines. The impact of pediatric CNS tumors on public health is small—there are fewer than 4000 cases diagnosed annually in the United States. CNS tumors represent 20% of all childhood malignancies.1 Most children with brain tumors will require RT during the course of their management. Based on the U.S. standard population data from 2000, which includes 78.8 million individuals aged birth to 19 years, annually there were 2742 cases of neuroepithelial tumors that included 654 cases of juvenile pilocytic astrocytoma, 118 cases of glioblastoma multiforme, 236 cases of ependymoma, and 433 cases of CNS embryonal tumors including medulloblastoma. Nonneuroepithelial tumors of interest to radiation oncologists included 197 cases of CNS germ cell tumors and 118 cases of craniopharyngiomas. These tumors require special care and remain a leading cause of cancer death in children.
Embryonal Central Nervous System Tumors
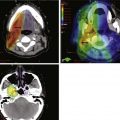
Embryonal tumors of the CNS are the most common malignant brain tumors in children. They are characterized by their infiltrative nature, aggressive patterns of growth, propensity for dissemination, and common need for intensive combined modality therapy. These tumors are found throughout the CNS and are further classified according to histologic and molecular features into two groups: primitive neuroectodermal tumors (PNET) and atypical teratoid rhabdoid tumors (ATRT). The PNET group includes two distinct entities—medulloblastoma (Fig. 65-1) and pineoblastoma (Fig. 65-2)—named for their site of origin in the posterior fossa and pineal region, respectively. ATRT are rare aggressive embryonal tumors that most commonly arise in young children in the brain and at peripheral soft tissue sites. With some exceptions, the diagnosis of ATRT is made on molecular grounds by demonstrating the deletion or mutation of the SMARCB1 gene found on chromosome 22q11.2.2 Atypical teratoid rhabdoid tumors unknowingly were considered the same as other PNET but are now known to carry a worse prognosis.
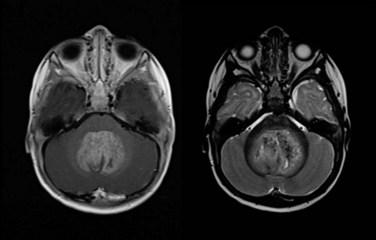
Figure 65-1 Medulloblastoma as shown on axial postgadolinium T1-weighted (left) and T2-weighted (right) MR images.
Medulloblastoma and supratentorial PNET are commonly diagnosed in young children. The relatively large numbers of patients and a cooperative spirit among caregivers have led to considerable understanding and progress in the treatment of these tumors. Patients are risk classified for treatment based on the three most important prognostic factors for tumor control— extent of disease, extent of resection, and histological subtype—and the most important prognostic factor for functional outcome—age at the time of irradiation. The radiation oncologist should be aware of the aggressive histologic variant of medulloblastoma (large cell or anaplastic medulloblastoma) that is more prone to dissemination through the cerebrospinal fluid (CSF).3,4 One also should be aware that there is a less aggressive variant (desmoplastic medulloblastoma) that may not require irradiation under certain conditions.
Medulloblastoma is a heterogeneous tumor and is now recognized as a number of different diseases based on specific molecular markers that have been retrospectively evaluated. Depending on the series, investigators have identified from four to six different subtypes based on the combination of molecular and classic histopathologic evaluation. A report by Northcott and associates5 described four distinct molecular variants of medulloblastoma, including WNT, SHH, and two non-WNT/SHH. The former two have clinical features that are associated with less aggressive phenotypes and the latter two, described as groups C and D in their report, have molecular and clinical features associated with more aggressive phenotypes, including overexpression of MYC and disseminated disease (group C) and the most common cancer isochromosome i17q and children between the ages of 4 and 15 years. One of the more striking findings by this group was the variation in molecular characteristics by age. Many of these findings have been confirmed by others groups,6 which suggests that tumor biologic information will be combined with clinical prognostic data to stratify patients in future trials.
At present, the treatment of embryonal tumors of the CNS is remarkably similar regardless of subtype or tumor location and consists of surgery, postoperative RT, and chemotherapy. The requirement of craniospinal irradiation, regardless of extent of disease, is the element that differentiates the treatment of these tumors. Approximately one third of patients present with neuraxis dissemination at the time of diagnosis as determined by magnetic resonance imaging (MRI) of the brain and spine (Fig. 65-3) or CSF cytology.7 Because the negative predictive value of these staging procedures remains relatively low, and the most common mode of failure remains distant, craniospinal irradiation is a mainstay in the treatment of these tumors with few exceptions.8
The side effects attributed to RT have been a primary concern for patients with medulloblastoma. The concern has been greatest for patients with average-risk disease, for whom long-term disease control is likely and the side effects of therapy have a lasting impact. The treatment of this tumor is technically demanding and consists of craniospinal irradiation and boost treatment of the anatomic posterior fossa. Until recently, the standard of care for an average-risk patient was postoperative craniospinal irradiation (CSI) to 36 Gy and boost treatment to 54 Gy using conventional fractionation of 1.8 Gy/day. In a cooperative group trial the CSI dose was lowered to 23.4 Gy for average-risk patients in a randomized comparison with 36 Gy CSI. This trial showed an increased risk of relapse in patients receiving the reduced CSI dose.9 Multiple-agent chemotherapy combined with reduced-dose CSI was found to be equivalent to standard-dose CSI and was subsequently adopted as a treatment standard for average-risk patients.10 Cognitive impairment continues to be a significant problem, with intelligence quotient (IQ) loss most commonly seen in children younger than the age of 8 years and those treated for high-risk medulloblastoma in which high-dose craniospinal irradiation (36 Gy) is required.11
The most recently completed Children’s Oncology Group study sought to define the best chemotherapy regimen (lomustine [CCNU], cisplatin, and vincristine vs. cyclophosphamide, cisplatin, and vincristine) to accompany low-dose craniospinal irradiation and the standard posterior fossa boost. No differences were observed in disease control (3-year event-free survival >83% in both arms); however, patients treated with the cyclophosphamide-containing regimen appeared to have more severe toxicity (18% vs. 25%).12 Attention has now turned to combining the two chemotherapy regimens and randomizing patients between 18.0 Gy and 23.4 Gy CSI followed by a second randomization between conventional boost treatment and treatment of less than the entire posterior fossa in an ambitious four-arm randomized trial developed for pediatric patients between the ages of 3 and 8 years. For those older than 8 years, the CSI dose remains at 23.4 Gy. The trial also includes a randomized comparison of conventional posterior fossa boost and boost treatment of the tumor bed using a 1.5-cm clinical target volume (CTV) margin.
Treatment of less than the entire posterior fossa after 23.4 Gy of CSI using focal radiation delivery techniques appears to be as effective as treatment of the entire posterior fossa based on recent trial results from St. Jude Children’s Research Hospital and participating centers involving 84 patients. In this prospective trial the cumulative incidence of posterior fossa failure was only 6.3% at 3 years.13 Treatment of these patients included postoperative CSI to 23.4 Gy, conformal posterior fossa boost to 36 Gy, and focal treatment of the tumor bed with a 2-cm margin. Chemotherapy was administered after RT. Patients on this study experienced a dose reduction to the anatomic posterior fossa of approximately 13%. The risks associated with reducing the CSI dose and limiting the boost treatment to the postoperative tumor bed with a limited margin must be balanced against the observed effects of RT on cognition, neurologic function, and growth and development.
Hyperfractionation has been tested in Europe to reduce toxicity.14 Although these investigations have shown that hyperfractionation may be isoeffective, lacking are the data that indicate side effects are reduced by this strategy. Considering logistics including the need for anesthesia in this often young patient population, investigators demand some certainty that there would be a benefit associated with hyperfractionated treatment before planning future trials.
Current cooperative group trials for very young children (<3 years of age) with medulloblastoma highlight the importance of RT in the treatment of these tumors. The recently completed Children’s Oncology Group protocol for children younger than the age of 3 years with localized medulloblastoma included 20 weeks of combination chemotherapy followed by second surgery when indicated and sequential irradiation of the posterior fossa and primary site before 20 additional weeks of maintenance chemotherapy.15 CSI was been omitted for these patients, and the doses to the posterior fossa (18 and 23.4 Gy) and primary site (45 to 54 Gy) were prescribed based on patient age, response, and risk status. A similar strategy was used in the Pediatric Brain Tumor Consortium (PBTC) protocol for similar patients with one exception: intrathecal chemotherapy (mafosfamide) was given during the first 20 weeks of chemotherapy as a substitute for CSI.16 Both protocols included RT after 20 weeks based on results from prior trials that showed a median time to progression of approximately 24 weeks.17,18 The targeting guidelines used in these studies were largely empirical: investigators attempted to minimize the dose to normal tissues while maintaining an acceptable rate of local tumor control.
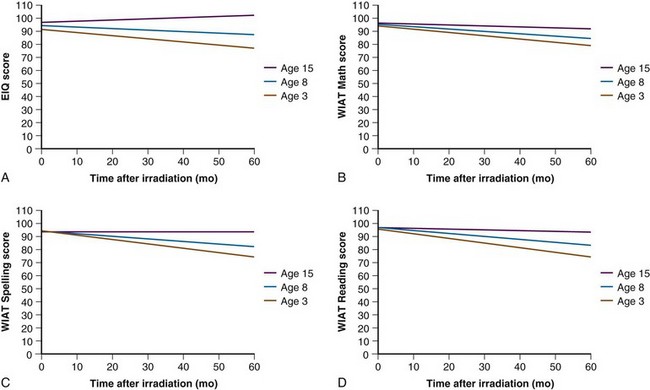
Fear of cognitive deficits has been the driving force in the design of current treatment trials for this young patient population. Age has the greatest impact on cognitive outcome after irradiation (Fig. 65-4). For very young children with medulloblastoma, a persistent decline in cognitive function has been observed by Walter and colleagues.19 The decline is not surprising given the median cranial dose of 35.2 Gy for patients with a median age of 2.6 years and the additive effects of tumor, surgery, and chemotherapy. Because a significant decline has also been observed for older children with medulloblastoma treated with lower craniospinal doses,11 CSI for the youngest children no longer appears to be a reasonable treatment option. Data concerning the effectiveness of yet lower CSI doses are extremely limited.20 These findings form the basis of the most recent trials for very young children. CSI has been omitted with the hope that the side effects of treatment will be reduced enough to provide for a meaningful quality of life among long-term survivors. There is a move to increase the age at which CSI is considered acceptable from age 3 years to 5 years at the time of treatment. Investigators acknowledge that they are trading fewer side effects for an increase in the rate of disseminated failure.
Ependymoma
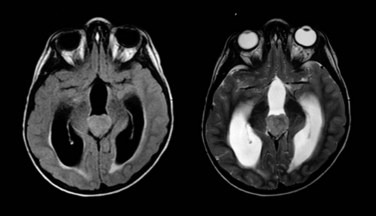
Ependymoma is the third most common CNS tumor in children, affecting approximately 236 individuals younger than the age of 19 years annually in the United States.1 It shares many of the clinical characteristics of more common gliomas, embryonal tumors, and less common germ cell tumors. Ependymoma may occur anywhere within the CNS and has the highest prevalence within the posterior fossa, arising from the floor or roof of the fourth ventricle or cerebellopontine angle, where it is known to invade the brainstem or envelop cranial nerves or vascular structures. Ependymoma also arises within the parenchyma of the cerebral hemispheres in older patients and rarely in the spinal cord. The interval from the onset of symptoms to diagnosis may be influenced by the young age at the time of diagnosis, the perceived slow growth rate of this tumor, and the remitting signs and symptoms of increased intracranial pressure resulting from obstructive hydrocephalus (Fig. 65-5).
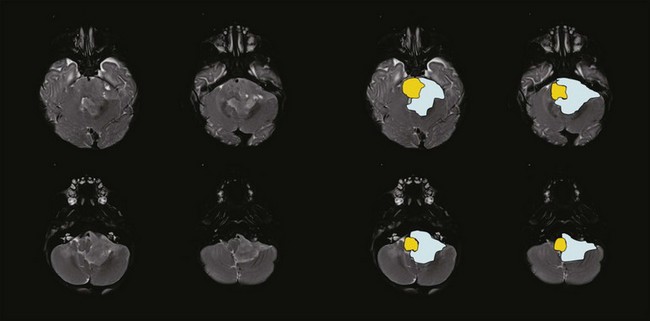
Ependymoma is most commonly diagnosed in children who are younger than 4 years of age. For the past two decades, progress in the treatment of this tumor has been slowed by concerns about late effects. Recent advances in neuroimaging, surgery, and RT have moved the field of neuro-oncology forward in the treatment of this tumor, resulting in more acceptable outcomes and providing new disease control benchmarks. Investigators treating these patients with contemporary surgery and RT have been able to increase event-free survivals measured at 3 years from 55% to 62% to 75% when measured at 7 years with remarkably limited treatment-related effects.21,22
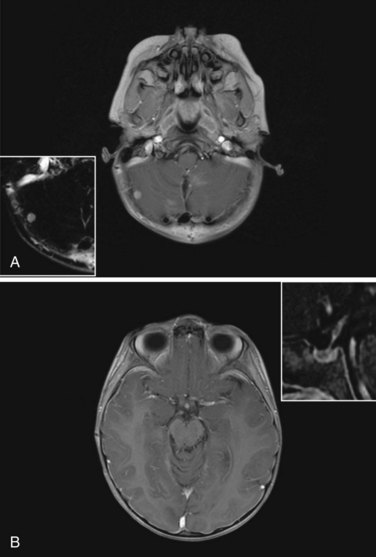
The standard of care for a child with localized ependymoma is to attempt gross-total resection, perform second surgery for potentially resectable residual tumor, and proceed with postoperative RT directed at the primary site using a 10-mm, anatomically confined CTV surrounding the residual tumor and/or tumor bed as defined on postoperative neuroimaging. The recommended total dose is 59.4 Gy using conventional fractionation because these tumors are prone to recur locally. Conventions regarding organs at risk apply; thus, for patients with tumors arising in the lower aspect of the posterior fossa, avoidance of the spinal cord and/or chiasm should be considered after approximately 54 Gy. Special considerations for patients with posterior fossa tumors may apply to very young children (generally those younger than the age of 18 months) with no evidence of residual tumor or patients who are noted to have signs and symptoms of severe brainstem injury after resection (postoperative seizure, hypertension, and ischemic changes on neuroimaging) who are at increased risk for necrosis. The recent trial of the Children’s Oncology Group showed that there was consensus to irradiate children younger than the age of 3 years and as young as 12 months postoperatively. The newest study from the Children’s Oncology Group, known as ACNS0831 and initiated in 2010, deploys a similar treatment algorithm and methods of irradiation with a smaller (5 mm) CTV margin. Children are randomized to receive four cycles of postirradiation chemotherapy (Fig. 65-6).
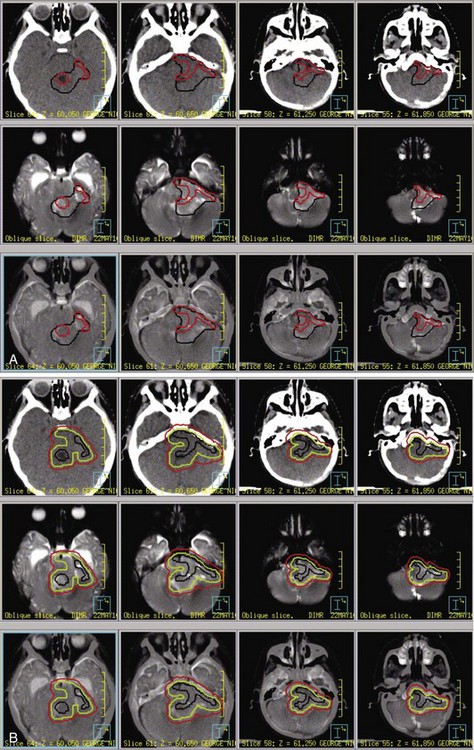
The prognosis for a patient with ependymoma is determined by tumor grade and extent of resection.23,24,25 Patients with overtly anaplastic tumors have inferior disease control when compared with those with differentiated tumors; however, the assignment of tumor grade can be difficult for those cases in which focal anaplasia occurs in the setting of a largely differentiated tumor. All patients should be treated with curative intent regardless of tumor grade. Extent of resection is an unequivocal prognostic factor: patients with substantial residual tumor have the worst prognosis. Because patients with varying amounts of residual tumor have achieved long-term survival after RT and modern surgical resection has altered the definition of near-total and subtotal resection, the volume of residual tumor differentiating between patients with good and poor prognosis remains unknown. It is incumbent on the radiation oncologist to question whether additional resection might be safely achieved in a patient with residual tumor referred for irradiation.
Research performed to evaluate molecular prognostic markers in ependymoma has revealed gain of 1q and homozygous deletion of CDKN2A as valuable predictors of poor survival in this disease26 and other markers associated with more favorable outcomes. Although molecular markers are not likely to supplant standard histopathologic evaluation, they will certainly contribute beneficial information for separating tumors that appear to have features of both differentiated and anaplastic ependymoma and determining which patients might require additional treatment after gross-total resection and irradiation.
High-Grade Astrocytoma
One study has shown a benefit for chemotherapy.27 This study included a randomization of 58 patients with high-grade astrocytoma to local irradiation versus local irradiation plus weekly vincristine followed by 12 month of carmustine, vincristine, and prednisone. The reported 5-year progression-free survival rate was 18% versus 46% favoring the use of chemotherapy, and patients with glioblastoma multiforme (n = 40) had a 5-year progression-free survival of 6% versus 42% favoring the use of chemotherapy. A larger study that included 172 patients randomized between local irradiation versus local irradiation plus preirradiation and postirradiation 8-in-1 chemotherapy (i.e., with vincristine, carmustine, procarbazine, cytarabine, hydroxyurea, cisplatin, dacarbazine, and methylprednisolone) showed no difference between the two study arms with 5-year progression-free survivals of 26% and 33%, respectively.28 This study was illustrative for the lack of concordance between institutional and expert pathology review. Thirty percent of patients in this study were found not to have high-grade glioma, and the survival estimates were in fact lower than originally reported.29


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


