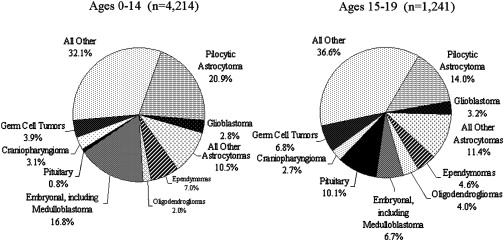
Central nervous system (CNS) tumors comprise 15% to 20% of all malignancies occurring in childhood and adolescence. They may present in a myriad of ways, often delaying diagnosis. Symptoms and signs depend on the growth rate of the tumor, its location in the central nervous system (CNS), and the age of the child. This article describes the presentation, diagnosis, and management of these tumors.
Central nervous system (CNS) tumors comprise 15% to 20% of all malignancies occurring in childhood and adolescence. Despite being relatively common, they only occur in between 2500 to 3500 children in the United States each year and may present in a myriad of ways, often delaying diagnosis. Symptoms and signs depend on the growth rate of the tumor, its location in the central nervous system (CNS), and the age of the child. Childhood brain tumors demonstrate greater histological variation, are more likely to be disseminated at the time of diagnosis, and more frequently are embryonal than those arising in adults.
The etiology for most childhood brain and spinal cord tumors is unknown. Specific syndromes are associated with a higher incidence of tumors. Patients who have neurofibromatosis type 1 (NF-1) have a higher incidence of low-grade gliomas, including visual pathway gliomas and other types of CNS tumors. Children who have tuberous sclerosis are prone to harbor giant-cell astrocytomas, and those who have the Li-Fraumeni syndrome have an increased predisposition to various different tumors including gliomas. Rarer conditions, such as the autosomally dominant inherited nevoid basal cell carcinoma syndrome (Gorlin syndrome) and the recessively inherited Turcot’s syndrome (germ line mutation of the adenomatosis polyposis coli gene) are associated with an increased incidence of medulloblastoma. Exposure to radiation therapy has been the only environmental factor consistently related to the development of brain tumors.
Presentation
Approximately one-half of all childhood brain tumors arise in the posterior fossa ( Table 1 ). The five major tumor types that arise subtentorially may present with focal neurologic deficits, but those filling the fourth ventricle are as likely to come to clinical attention because of obstruction of cerebrospinal fluid with associated hydrocephalus. The classical triad associated with increased intracranial pressure of morning headaches, nausea, and vomiting, may occur, but nonspecific headaches are more frequent. In infants, cerebrospinal fluid obstruction with dilatation of the third ventricle and the resultant tectal pressure causes paresis of upgaze may result in downward deviation of the eyes, the setting sun sign.
| Tumor | Relative Incidence | Presentation | Diagnosis | Prognosis |
|---|---|---|---|---|
| Medulloblastoma | 35% to 40% | 2–3 months of headaches, vomiting, truncal ataxia | Heterogeneous or homogeneously enhancing fourth ventricular mass; may be disseminated | 65% to 85% survival; dependent on stage/type; poorer (20% to 70%) in infants |
| Cerebellar astrocytoma | 35% to 40% | 3–6 months of limb ataxia; secondary headaches, vomiting | Cerebellar hemisphere mass, usually with cystic and solid (mural nodule) components | 90% to 100% in totally resected pilocytic type |
| Brain stem glioma | 10% to 15% | 1–4 months of double vision, unsteadiness, weakness, and other cranial nerve deficits, facial weakness, swallowing deficits, and other deficits | Diffusely expanded, minimally or partially enhancing mass in 80%; 20% more focal tectal or cervicomedullary lesion | 90% + 18-month mortality in diffuse tumors; better in localized |
| Ependymoma | 10% to 15% | 2–5 months of unsteadiness, headaches, double vision, and facial asymmetry | Usually enhancing, fourth ventricular mass with cerebellopontine predilection | 75% + survival in totally resected lesions |
| Atypical Teratoid/Rhabdoid | >5 (10% to 15% of infantile malignant tumors) | As in medulloblastoma, but primarily in infants; often associated facial weakness and strabismus | As in medulloblastoma, but often more laterally extended | 10% to 20% (or less) survival in infants |
The suprasellar and pineal regions are relatively frequent sites for supratentorial childhood brain tumors. Tumors in the suprasellar region, primarily craniopharyngiomas, visual pathway gliomas, and germinomas, may present with complex visual findings including unilateral or bilateral decreased visual acuity and hard to characterize visual field loss, as well as hormonal dysfunction. In the pineal region, various different tumor types may occur in the pediatric years, including germinomas, mixed germ cell tumors, pineoblastomas, and lower-grade pineocytomas. Pineal region lesions characteristically cause compression or destruction of the tectal region of the brain stem, and result in Parinaud’s syndrome, manifested by paralysis or paresis of upgaze, retraction or convergence nystagmus, pupils that react better to accommodation than light, and lid retraction. Most cortical childhood tumors are gliomas, usually low-grade, but they are anaplastic in approximately 20% of cases. Other tumor types (supratentorial primitive neuroectodermal tumors and ependymomas) may occur. Unlike the situation in adulthood, pediatric low-grade gliomas do not mutate frequently to higher-grade gliomas during childhood. In younger children, large benign supratentorial lesions, such as diffuse infantile gangliogliomas/gliomas and dysembryoplastic neuroepithelial tumors may be misdiagnosed as more aggressive lesions.
Diagnosis
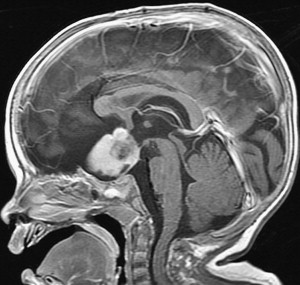
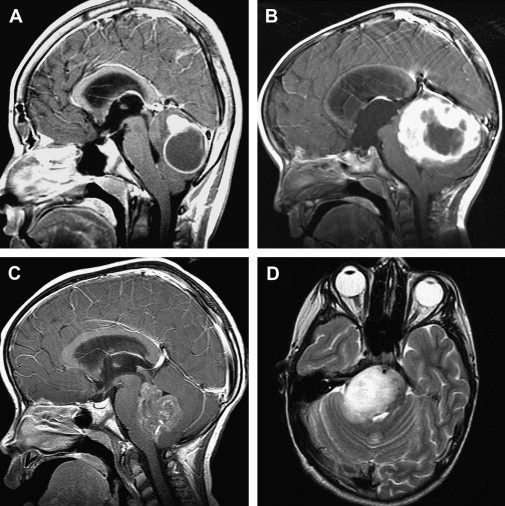
The diagnosis of pediatric brain and spinal cord tumors has been simplified by advances in neuroimaging. Because of the speed and availability of CT, it is often the first imaging technique obtained for children with suspected intracranial pathology and, if properly done, CT will detect 95% or more of brain tumors. Because of the superior image contrast of MRI, however, it is essential in the diagnosis of brain tumors, and its multiplanar capabilities offer far superior tumor localization. Based on clinical and neuroradiographic findings, brain tumors have characteristic presentations, especially those arising in the posterior fossa ( Fig. 1 ). Other MRI techniques such as magnetic resonance spectroscopy, which supplements anatomic findings with biochemical data and possibly, in the future, diffusion tensor imaging, especially tractography, may aid in characterizing the type of tumor and its anatomic interrelationships.
For the diagnosis of spinal cord tumors or determination of leptomeningeal dissemination of tumors, spinal MRI has supplanted all other techniques, including myelography or CT studies. In attempts to avoid postoperative artifacts, an MRI of the entire neurospinal axis often is undertaken before surgery in patients who have presumed malignant tumors.
In selected cases, positron emission tomography (PET) scanning may provide additional information, but it is usually most useful in supplying baseline diagnostic information, as a means to follow the tumor over time. PET is most helpful in the determination of transformation of a lower-grade tumor (primarily glial) to a higher-grade neoplasm and the separation of post-therapy, especially postradiation, treatment effects from tumor progression.
Diagnosis
The diagnosis of pediatric brain and spinal cord tumors has been simplified by advances in neuroimaging. Because of the speed and availability of CT, it is often the first imaging technique obtained for children with suspected intracranial pathology and, if properly done, CT will detect 95% or more of brain tumors. Because of the superior image contrast of MRI, however, it is essential in the diagnosis of brain tumors, and its multiplanar capabilities offer far superior tumor localization. Based on clinical and neuroradiographic findings, brain tumors have characteristic presentations, especially those arising in the posterior fossa ( Fig. 1 ). Other MRI techniques such as magnetic resonance spectroscopy, which supplements anatomic findings with biochemical data and possibly, in the future, diffusion tensor imaging, especially tractography, may aid in characterizing the type of tumor and its anatomic interrelationships.
For the diagnosis of spinal cord tumors or determination of leptomeningeal dissemination of tumors, spinal MRI has supplanted all other techniques, including myelography or CT studies. In attempts to avoid postoperative artifacts, an MRI of the entire neurospinal axis often is undertaken before surgery in patients who have presumed malignant tumors.
In selected cases, positron emission tomography (PET) scanning may provide additional information, but it is usually most useful in supplying baseline diagnostic information, as a means to follow the tumor over time. PET is most helpful in the determination of transformation of a lower-grade tumor (primarily glial) to a higher-grade neoplasm and the separation of post-therapy, especially postradiation, treatment effects from tumor progression.
Classification
Because of the histologic variability of childhood brain and spinal cord tumors, classification is often difficult and, at times, subjective. In most cases, diagnosis continues to be made predominantly upon light microscopy findings. In selective situations, such as in embryonal tumors, especially atypically teratoid/rhabdoid lesions, immunohistochemistry has aided diagnosis greatly. Although the molecular underpinnings of childhood brain tumors increasingly have been unraveled, molecular techniques have not been incorporated extensively into most classification systems. Evaluation of the mitotic activity of the tumor, assessed by mitotic indices, usually does not change classification, but may be helpful, in selected situations, in determining prognosis and approach to therapy.
Specific tumor types
Discussions of the biology of the tumor, its growth pattern, management, and prognosis are discussed best within individual tumor types. For most tumors, the same modalities of treatment are used (ie, surgery, radiation, and in an increasing number of patients chemotherapy), but the use of each of these types of treatment is not only dependent on the type of tumor present, but also on its location in the CNS tumor and the age of the child. Biologic therapies are just being introduced into management, and to date, have been reserved primarily for those patients who fail initial treatment.
Medulloblastoma
Medulloblastoma, which by definition arises in the posterior fossa, is the most common malignant brain tumor of childhood ( Fig. 2 A). Medulloblastomas usually are diagnosed in children less than 15 years of age, and they have a bimodal distribution, peaking at 3 to 4 years of age and then again between 8 and 9 years of age. For unknown reasons, there is a male predominance. Ten percent to 15% of patients are diagnosed in infancy. The classical, or undifferentiated, type of medulloblastoma, comprising 70% or more of medulloblastomas, is composed of densely packed cells with hyperchromatic, round, oval, or carrot-shaped nuclei and minimal cytoplasm. The large-cell, or anaplastic variant, which has pleomorphic nuclei, prominent nucleoli and more abundant cytoplasm, as well as possibly higher mitotic and apoptotic indices, increasingly has been recognized and may carry a poorer prognosis. By contrast, the desmoplastic, at times nodular, medulloblastoma variant seems more responsive to therapy and may have a better prognosis.
Biology
Medulloblastoma is thought to originate from a primitive cell type in the cerebellum, arising from one of the two cerebellar germinal zones, the ventricular zone that forms the innermost boundary of the cerebellum or the external germinal layer that lines the outside of the cerebellum. The multipotent progenitor cells of the ventricular zone have been postulated to be the primary site of development of classical medulloblastomas, and the classical tumor is more likely to express more primitive, possibly stem cell markers. Medulloblastomas arising from the external granular layer on the other hand are believed to originate from a more neuronally restricted granular cell precursor, and are more likely to be desmoplastic and express markers of granular cell lineage.
Various genes and signaling pathways have been identified as active in medulloblastoma and support progenitor cell theories. The nevoid basal cell carcinoma syndrome, which is caused by an inherited germ line mutation of the PTCH gene on chromosome 22, encodes the sonic hedgehog (SHH) receptor PATCHED1 (PTC1), which normally represses SHH signaling. Somatic mutation of PTC1 has been associated predominantly with the desmoplastic variant, possibly from external granular layer precursors, and this pathway is likely a potential therapeutic target for 10% to 20% of medulloblastomas. Classical medulloblastomas are less likely to have abnormalities in the SHH pathway and may be more likely to arise from the internal granular layer. Another signaling pathway that has been identified in a subset of patients with medulloblastoma has been the WNT pathway, which is aberrant in Turcot’s syndrome. Patients who have this molecular abnormality, which has been noted in as many as 15% of patients and may affect growth and survival of multipotential cerebellar progenitor cells, have been demonstrated to have better prognosis.
Specific molecular genetic abnormalities have been associated with medulloblastoma and variably correlated with survival. Amplification of the MYCC oncogene has been associated with the large-cell variant and poorer outcome. Similarly, expression of the tyrosine kinase receptor ERBB2 has been demonstrated in 40% of medulloblastomas and is also predictive of poor outcome. Increased expression of the neurotrophin-3 receptor (TRKC), which regulates proliferation, differentiation, and granular layer cell death, has been associated with better survival. Amplification of the OXT2 homeobox gene, a retinoid target, has been identified in the anaplastic medulloblastoma variant. Gene expression profiling has demonstrated differences in metastatic and nonmetastatic tumors, as platelet-derived growth factor receptor beta and members of the RAS-MAP kinase pathway are up-regulated significantly in metastatic tumors.
These and other biologic alterations have allowed a significant better understanding of medulloblastoma. In time, it is likely that they will be incorporated into staging schema for medulloblastoma and also will act as therapeutic targets. To date, however, medulloblastomas predominantly are staged and treated based on clinical parameters.
Management
In most patients who have medulloblastomas, the initial step in treatment is surgical resection. Total or near-total resection of the primary tumor site has been correlated with better survival, predominantly in nondisseminated patients. Such resections will result in avoidance of permanent ventriculoperitoneal cerebrospinal drainage in over 60% of patients. Significant postoperative complications may occur, including both septic and aseptic meningitis, postoperative cerebrospinal fluid leaks, and increased neurologic morbidity caused by direct cerebellar or brain stem damage. The cerebellar mutism syndrome has been identified in up to 25% of patients following resection of midline cerebellar tumors. This syndrome presents as the late (delayed) onset of mutism associated with a variable constellation of nystagmus, truncal hypotonia, dysmetria, dysphagia, other supranuclear cranial nerve palsies, and marked emotional lability. The neurophysiologic mechanism underlying this syndrome is unclear, but it is believed to be related to vermian damage and possibly impaired dentatorubrothalamic connections to the supplementary motor cortex. Symptoms may persist from weeks to months and approximately 50% of those affected will have significant sequelae 1 year after surgery. Neurosurgery-related complications have not been related clearly to more aggressive surgery.
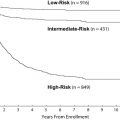
Following surgery, patients usually are stratified into one of two risk groups, based on extent of surgical resection and disease extent at the time of diagnosis ( Table 2 ). Neuroradiographic staging, although critical, remains problematic, as central review of international studies has demonstrated inadequate spinal neuroimaging or misinterpreted studies in nearly 25% of patients. Adequate staging requires meticulous, multiplanar neuro-axis imaging and lumbar cerebrospinal fluid analysis. In time, as these risk groups are modified by the inclusion of other factors, including histological features and molecular genetic parameters, an intermediate risk group of patients may become more apparent (see Table 2 ).
| Average-Risk | High-Risk | |
|---|---|---|
| Tumor extent | Localized | Disseminated |
| Tumor resection | Total; near total | Subtotal; biopsy |
| Histology | Classical; desmoplastic/nodular | Anaplastic/large cell |
| Biologic parameters | Neurotrophin-3 receptor expression; sonic hedgehog lineage markers | ↑ MYCC amplification; ↑ ERBB2 expression; OXT2 amplification |
Patients greater than 3 years of age with average-risk disease are treated conventionally with craniospinal (2400 cGy) and local boost radiotherapy (5580 cGy), supplemented with adjuvant chemotherapy. The dose of craniospinal radiation therapy for children who have nondisseminated disease has been decreased by one-third (from 3600 cGy) with maintained efficacy, as long as chemotherapy is given during and after radiotherapy. Studies are underway attempting to determine if a further reduction of the dose of craniospinal radiotherapy from 2340 cGy to 1800 cGy will result in equivalent survival figures and better neurocognitive outcome.
Different chemotherapeutic regimens have shown benefit in medulloblastoma. Probably the best tested is the use of vincristine during radiotherapy and the combination of CCNU, cisplatin and vincristine, or cyclophosphamide, cisplatin, and vincristine following radiotherapy. Another approach, demonstrating similar survival, has used essentially the same agents in a truncated, higher-dose fashion supported by peripheral stem cell rescue. With such combination approaches used during and after radiotherapy, over 80% of children with average-risk medulloblastoma are alive and free of disease five years following diagnosis, most of whom are apparently cured from their disease. The use of preradiotherapy chemotherapy has resulted in inferior survival.
Children older than 3 years who have high-risk medulloblastoma have approximately a 50% to 60% 5-year disease-free survival after treatment with higher doses of craniospinal radiation therapy (3600 cGy) and similar doses of local radiotherapy, as used for children with average-risk disease, and chemotherapy during and after radiation therapy. Recent trials have included the use of carboplatin as a radiosensitizer during radiation therapy and the delivery of higher-dose chemotherapy, essentially an intensified cisplatin, cyclophosphamide, vincristine, and etoposide regimen, supported by peripheral stem cell rescue, following radiotherapy, with possibly better results. The added efficacy of biologic therapy, such as retinoic acid and tyrosine kinase inhibitors, is being explored in this subset of patients.
Treatment of children younger than 3 years of who have medulloblastoma is problematic. Because of the immaturity of the brain and the resultant deleterious effects of whole brain irradiation on the very young child, there is significant reluctance to use craniospinal radiation therapy. Management of infants is complicated further by the increased likelihood of dissemination at the time of diagnosis in younger patients, because as many as 40% of children younger than 3 years who have medulloblastoma will have disseminated disease at diagnosis. Although various chemotherapeutic approaches have been used, and others are under active study, the most important predictor of outcome is likely not the type of regimen employed, but rather the biology of the tumor. Infants who have desmoplastic/nodular tumors are quite responsive to chemotherapy, and 75% or greater of patients harboring this histologic variant may be cured by chemotherapy alone. Outcome is less favorable in infants who have classical, undifferentiated medulloblastoma, especially in those who have disseminated disease at the time of diagnosis. More intensive chemotherapeutic regimens using peripheral stem cell support or regimens that have been supplemented with high-dose, intravenous, and intrathecal methotrexate have shown possible increased efficacy. The safety and efficacy of focal radiation therapy to the primary tumor site also is being explored in this age group.
Children of all ages with medulloblastoma who survive are at risk for significant long-term sequelae. The whole brain portion of craniospinal radiation therapy has been implicated as the primary cause of long-term neurocognitive deficits. Other factors, however, may play a significant role in sequelae encountered. These include: the tumor’s location extent and the presence of hydrocephalus at diagnosis, postsurgical complications, the age of the patient, the potential deleterious effects of concomitant chemotherapy, and the additive toxicity of local radiotherapy. Neurocognitive difficulties are the most common sequelae seen, and even after reduction of whole-brain radiotherapy from 3600 cGy to 2340 cGy, most children, especially those younger than 7 years of age, will have significant intellectual difficulties. Deficits include demonstrable post-treatment drop in overall intelligence, as well as deficits in perceptual motor ability, memory tasks, verbal learning, and executive function. Moderate-to-severe learning difficulties are common.
Neuroendocrine sequelae are also relatively common, but these seem to be somewhat less frequent in those children who have been treated with reduced doses of craniospinal radiation therapy. Of the endocrinologic sequelae, growth hormone insufficiency is the most common, occurring in almost all prepubertal children who have received 3600 cGy of craniospinal radiation and probably 50% or more of those children receiving 2340 cGy of whole-brain irradiation therapy. Thyroid dysfunction also occurs frequently and is caused not only by the dose of radiotherapy delivered to the hypothalamic region, but also by scatter radiation to the thyroid gland.
Permanent neurologic sequelae, including motor difficulties, sensory dysfunction, hearing impairments caused by the tumor, chemotherapy (especially cisplatinum), or radiation therapy, and visual abnormalities increasingly have been recognized in long-term survivors. Late-onset cerebrovascular damage resulting in stroke-like episodes is not uncommon, and can cause devastating long-term complications. Survivors are at risk for small vessel microangiopathy and more subtle vascular-related sequelae, such as seizures. Secondary tumors are another long-term complication of medulloblastoma and may be related to the underlying genetic predisposition of these patients to tumors and/or to the radiotherapy employed. Both meningiomas and gliomas can occur at any time following successful treatment, and, in general, high-grade gliomas tend to predominate in the first 5 to 10 years following therapy, while the risk of meningiomas continues to increase over time.
Supratentorial Primitive Neuroectodermal Tumors
Supratentorial primitive neuroectodermal tumors are characterized by undifferentiated or poorly differentiated neuroepithelial cells that may show some degree of differentiation. Although similar histologically, they are biologically different from medulloblastomas. Various different names, including cerebral neuroblastomas, have been used for these tumors, which, by definition, must occur above the tentorium, primarily in the cerebral cortex and less frequently in the diencephalic region. They are infrequent, comprising 2.5% of all childhood brain tumors. The tumors are staged based predominantly on tumor extent at diagnosis, although approximately 20% or less of lesions have evidence of dissemination.
The degree of surgical resection has been related variably to outcome. Postsurgical management has been similar to that employed for high-risk medulloblastoma patients, with most children being treated with craniospinal and local boost radiotherapy and aggressive adjuvant chemotherapy. The need for craniospinal radiation therapy has never been demonstrated clearly, although it is used frequently. Reported 5-year progression-free survival rates have ranged from 30% to 60%, with most series finding that approximately 50% of affected patients survive.
Pineoblastomas
Although pineoblastomas are classified as pineal parenchymal tumors, they are conceptualized most commonly as a subvariant of embryonal tumors and are managed similarly to high-risk medulloblastomas. They represent approximately 25% of tumors that occur in the pineal region. Dissemination at the time of diagnosis is present in 20% to 30% of patients.
Total resections before the initiation of adjuvant treatment are uncommon because of tumor location. Survival figures after treatment with craniospinal plus local boost radiation therapy and adjuvant chemotherapy, similar to that being used for high-risk medulloblastoma patients, have been quite variable, with some studies reporting relatively optimistic 5-year progression-free survival rates of as high as 60%. Other series have reported much less favorable outcomes, especially in very young children who are not treated with craniospinal radiotherapy.
Atypical Teratoid/Rhabdoid Tumors
Atypical teratoid/rhabdoid tumors (AT/RTs) first were recognized as a discreet entity in the late 1980s. These lesions, which predominantly occur in children younger than 3 years, but may be first diagnosed in older children and adolescents, histologically are characterized by rhabdoid cells intermixed with a variable component of primitive neuroectodermal, mesenchymal, and epithelial cells. The rhabdoid cell is a medium-sized round-to-oval cell with distinct borders, an eccentric nucleus, and a prominent nucleolus. The primitive neuroectodermal component of AT/RTs is indistinguishable from that found in other forms of primitive neuroectodermal tumors. Immunohistochemical studies demonstrated that AT/RTs were different from medulloblastomas, because the rhabdoid component of the tumor characteristically stained positive for epithelial membrane antigen, vimentin, cytokeratin, glial fibrillary acidic protein, and, at times, smooth muscle actin and neurofilament protein. Molecular genetic studies have demonstrated that AT/RTs are distinct from other embryonal tumors and are characterized by deletions or mutations of the tumor suppressor gene hSNF5/INI1 located in the chromosomal region 22q11.2.
Management of AT/RTs has been extremely problematic. The tumor arises equally in the posterior fossa or supratentorium. Dissemination has been reported in approximately 25% of patients at diagnosis. Outcome after treatment of infants on protocols used for children younger than 3 years with medulloblastoma, including high-dose chemotherapy protocols, has been disappointing, with prolonged survival occurring in less than 20% of patients who had nondisseminated tumors, primarily in those who had undergone a total or near-total resection. Various different chemotherapeutic approaches are under study, including adding methotrexate to the drug regimen or using protocols that are hybrids of the infant medulloblastoma and sarcoma treatment regimens. Survival seems more favorable in patients older than 3 years at diagnosis treated with extensive resections, craniospinal and local boost radiotherapy, and chemotherapy.
Gliomas
High-grade gliomas
These tumors present most frequently between 5 and 10 years of age. Patients may present with headaches, motor weakness, personality changes and seizures; however, seizures are more typical of low-grade cortical lesions. On CT and MRI, high-grade gliomas (HGG) typically appear as irregularly shaped lesions with partial contrast enhancement and peritumoral edema with or without mass effect.
Radical (greater than 90%) surgical resection is the most powerful predictor of favorable outcome in HGG when followed by irradiation. Only 49% of tumors in the superficial hemisphere and 8% of tumors in the midline or deep cerebrum are amenable to radical resection, however. Local or wide-field irradiation to 5000 cGy to 6000 cGy is the mainstay of therapy. The addition of radiation therapy has improved 5-year survival rates (10% to 30%) compared with surgery alone (0%). Although initial reports demonstrated a benefit of adjuvant chemotherapy with prednisone, CCNU, and vincristine (pCV) compared with radiotherapy alone (46% versus 18%), a subsequent trial comparing pCV with the eight-in-one regimen failed to show the same benefit (26%). A review of the histology from the original trial with pCV suggested that a significant portion of patients (69/250) actually did not meet the central consensus definition of HGG. Most recently, temozolamide and concurrent radiation followed by maintenance temozolomide therapy has been used; however this regimen has shown no improvement in survival. To date, no large randomized clinical trial has demonstrated a benefit of chemotherapy clearly. High-dose chemotherapy for HGG has shown effective responses, and despite significant associated toxicity, may warrant further investigation.
Biologic therapy, such as drugs that target angiogenesis, is being investigated as an alternative approach. Specific biologic therapeutic targets have not been defined well for childhood HGG. For example, although 80% of pediatric HGG overexpress the EGFR protein, amplification of the EGFR gene is rare compared with EGFR amplification in one-third of adult glioblastomas. A more recent expression profiling study of childhood HGG revealed increased expression of the EGFR/HIF/IGFBP2 pathway. The TP53 gene is mutated in 34% of HGG in children younger than 3 years and only 12% of HGG in children older than 3 years, while the 5-year progression-free survival in those with low expression of p53 protein is 44% compared with 17% in those with high expression. Deletions for p16INK4a and p14ARF of the Rb pathway are observed in only 10% of pediatric HGG. These data indicate that the development of pediatric HGG may follow different pathways from the primary or secondary paradigm of adult glioblastomas and as such may require differently tailored biologic therapy than is being employed for adults with HGG.
Low-grade gliomas
Most low-grade cortical gliomas in children are juvenile pilocytic astrocytoma (JPA) or diffuse fibrillary astrocytoma. Other forms, such as oligodendroglioma, oligoastrocytoma and mixed glioma are much less common. Low-grade cortical gliomas (LGG) most commonly present with headache and seizure. On CT, diffuse astrocytomas appear as ill-defined, homogeneous masses of low density without contrast enhancement. MRI usually shows a mass that is hypodense on T1 weighted and hyperintense on T2 weighted images with little enhancement. Imaging of JPA lesions is similar to the cerebellar counterpart.
Complete surgical resection is curative for most, and even with incomplete excision, long-term progression-free survival is common. If subsequent progression occurs, then re-resection generally is undertaken. For patients who have progressive disease not amenable to resection, irradiation with 5000 cGy to 5500 cGy is warranted. Chemotherapy is reserved for very young children and infants and most commonly includes carboplatin and vincristine.
Overall 5-year survival is 95%, while progression-free survival is 88%. Less favorable results have been seen in patients who have nonpilocytic astrocytoma.
Chiasmatic gliomas
Gliomas of the visual pathway, which also may extend to the hypothalamus and thalamus, comprise a relatively common form of childhood glioma. Tumors of the optic chiasm are usually low-grade. Twenty percent of children who have (NF-1) will develop visual pathway tumors, predominantly JPA, during childhood. Visual pathway tumors may cause visual loss, strabismus, proptosis and/or nystagmus. Extension to the hypothalamus may present with endocrinologic disturbances, including precocious puberty. Imaging demonstrates similar characteristics of low-grade gliomas that present elsewhere ( Fig. 3 ). Optic pathway lesions have limited capacity to spread, as they are confined to migrate between the optic nerve and chiasm.