Neoplastic transformation in the nervous system is a multistep process in which the normal controls of cell proliferation and cell-cell interaction are suppressed or disabled. This process involves the alteration of several types of genes, including oncogenes, tumor suppressor genes, DNA repair genes, and cell death genes, among others.1 Our increased knowledge of the molecular genetics that underlie this process has been facilitated by improved methods for detailed and rapid analysis of molecular characteristics of tumors, and information obtained from the application of current technology is beginning to influence the clinical diagnosis and management of CNS cancer. Results from comprehensive analyses of tumor genomes (DNA, DNA modification) and transcriptomes (mRNA, miRNA) are proving especially powerful with regard to applications involving the differential diagnosis of adult malignant gliomas,2,3,4,5,6,7 embryonal brain tumors such as medulloblastoma,8,9 and to the subset of meningiomas that display variable clinical course.10 In addition to clinical applications, detailed molecular characterizations of CNS tumors continue to help improve our ability to develop increasingly accurate and relevant mouse models of brain tumors,11 which, in turn, facilitate precise dissection of tumorigenic pathways investigation of therapy-response relationships,12,13 and improved understanding of the earliest stages of brain tumorigenesis, including the nature of cells that give rise to brain tumors.
NEUROLOGIC TUMOR SYNDROMES
In addition to information obtained through molecular characterizations of sporadically occurring CNS tumors, as well as through cancer stem cell investigations, much of our current understanding of brain tumorigenesis is associated with decades of observation and analysis of inherited cancer predisposition. Neurologic tumor syndromes are accompanied by characteristic panoplies of both neurologic and nonneurologic tumors. A catalog of the major primary brain tumors associated with neurologic tumor syndromes would feature optic nerve gliomas and other astrocytomas in neurofibromatosis type 1 (NF1), ependymomas and meningiomas in neurofibromatosis type 2 (NF2), various malignant gliomas in Li-Fraumeni syndrome, Turcot syndrome and the hereditary glioma pedigrees, and medulloblastomas in Gorlin, Turcot, and Li-Fraumeni syndromes.14
Linkage studies that were applied to the initial chromosome regional assignments of the genes associated with these tumor syndromes15 revealed the NF1 gene as residing on chromosome 17q, the NF2 gene on chromosome 22q, and at least one gene for the Turcot syndrome on chromosome 5q (APC gene). For the Li-Fraumeni syndrome, mutation analyses identified the TP53 gene on 17p and the hCHK2 gene on 22q as being responsible for cancer predisposition. Similarly, germ line mutations of the chromosome 10q-localized PTEN gene are responsible for the multicancer Cowden syndrome, and the inactivation of PTEN is very common among high malignancy grade astrocytomas in adults.
As has been the case for the examination of tumor DNAs, studies of constitutional DNAs from brain tumor patients for the identification of inherited allelic variants that increase the likelihood of brain tumor occurrence have evolved toward comprehensive investigation, as exemplified by genome-wide association study approaches that have recently revealed possible glioma susceptibility loci. Two recent reports, published simultaneously and independently by multi-institutional consortia, suggested susceptibility loci at 9p21 (CDKN2A-CDKN2B as candidate loci) and 20q13 (RTEL1 as candidate locus reviewed in refs. 16 and 17). Consequently, there appear to be additional CNS tumor predisposing or susceptibility genes, not necessarily associated with definable clinical syndromes, that increase risk for CNS tumor development, and our knowledge of such factors is rapidly expanding from such investigations.
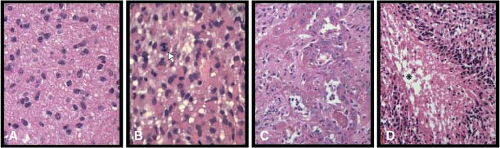
FIGURE 28.1 Examples of grade II (panel A), grade III (anaplastic: panel B), and grade IV (glioblastoma: panels C and D) astrocytoma histopathology. Arrow in panel B shows mitotic figure that is a classification criterion of grade III malignancy. Panels C and D show microvascular proliferation and necrosis with perinecrotic cellular palisading, respectively, which are diagnostic criteria of glioblastoma, grade IV. The asterisk in panel D denotes the necrotic focus.
CNS TUMOR HISTOPATHOLOGY AND MOLECULAR CORRELATES
Diffuse, Fibrillary Astrocytomas
Diffuse, fibrillary astrocytomas are the most common type of primary brain tumor in adults. These tumors are divided histopathologically into three grades of malignancy14: World Health Organization (WHO) grade II diffuse astrocytoma, WHO grade III anaplastic astrocytoma, and WHO grade IV glioblastoma (GBM) (Fig. 28.1). WHO grade II diffuse astrocytomas are the most indolent of the spectrum. Nonetheless, these tumors are infiltrative (Fig. 28.1A) and have a marked potential for malignant progression.18
TABLE 28.1 COMMON CENTRAL NERVOUS SYSTEM TUMORS AND CORRESPONDING GENE ALTERATIONSa
Common Adult Tumors
Frequent Gene and Chromosomal Alterations
Grade II astrocytoma
IDH1, TP53
Grade III anaplastic astrocytoma
IDH1, TP53-MDM2/4,CDKN2A-CDK4/6-RB
Grade IV glioblastomas
TP53-MDM2/4, CDKN2A-CDK4/6-RB,EGFR, PTEN, NF1
Grade II oligodendroglioma
IDH1, chromosome 1p-19q translocations
Grade III oligodendroglioma
IDH1, chromosome 1p-19q translocations
Meningioma
NF2
Common Pediatric Tumors
Frequent Gene and Chromosomal Alterations
Medulloblastoma
PTCH,MYCC,MYCN, chromosome 17p deletions
Ependymoma
NF2 (spinal), chromosome 22 deletions (central)
Pilocytic astrocytoma
KIAA1549-BRAF fusion rearrangements
aOncogene alterations are in bold text; tumor suppressor gene alterations are in plain text; functionally related gene alterations have been grouped (e.g., TP53-MDM2/4).
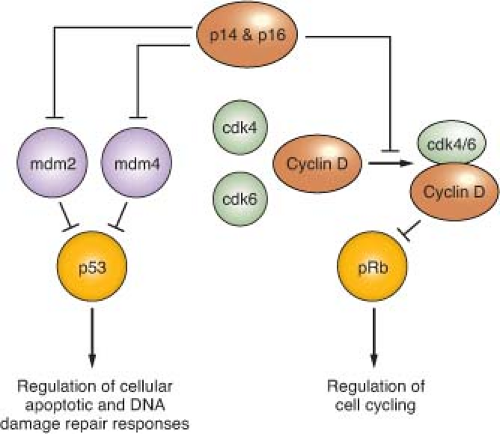
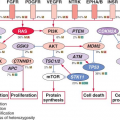
Alterations of p53, a tumor suppressor encoded by the TP53 gene on chromosome 17p, play a key role in the development of at least one-third of all three grades of adult astrocytoma.19,20 In addition, in higher-grade astrocytomas, p53 function may be deregulated by alterations of other genes, including amplification of MDM2 or MDM4 and 9p deletions that result in loss of the p14 product of the CDKN2A gene (Fig. 28.2). A recent survey of this pathway demonstrated gene alterations that compromise p53 function in 87% of glioblastoma.21
Studies revealing frequent alterations of TP53 in sporadic astrocytoma are complemented by various model system investigations that support the contribution of p53 inactivation in the early stages of astrocytoma formation. For instance, cortical astrocytes from mice without functional p53 appear immortalized when grown in vitro and acquire a transformed phenotype with sustained propagation in defined media.22 Cortical astrocytes from mice with haploid TP53 status behave more like wild type astrocytes and only show signs of immortalization and transformation after losing their sole wild type copy of TP53.22 Results associated with p53 inactivation in genetically modified mouse models also support the importance of p53 loss of function in promoting astrocytoma initiation, although such demonstrations have been reported in the context of an accompanying, second gene alteration, such as NF1 gene inactivation.23 In total, there is ample evidence provided through numerous avenues of investigation that indicate the importance of compromised p53 function to the formation of astrocytoma.
FIGURE 28.2 Regulation of p53 and pRb function. p14 and p16 function is inactivated in approximately half of glioblastoma, as well as in a significant fraction of grade III (anaplastic) astrocytoma, due to homozygous deletion of a DNA sequence at chromosomal location 9p21 that encodes each of these tumor suppressors. The genes encoding mdm 2 and 4, as well as for cdk4 and 6, are amplified in some high-grade astrocytomas, and provide alternative genetic mechanisms to the p14 + p16 gene deletions for achieving suppression of p53 and pRb function. The TP53 and RB genes that encode these tumor suppressor proteins are themselves inactivated in many high-grade astrocytomas, and in such instances the gene alterations affecting upstream regulators are not observed. Proteins indicated in green are oncogenic, whereas those indicated in red act as negative regulators of cell growth (tumor suppressors). Percent values indicate gene alteration frequencies, as defined by The Cancer Genome Atlas project.21
During the 16 months prior to this writing, it has become well established that mutations of the isocitrate dehydrogenase 1 gene (IDH1) occur in a large fraction of grade II and grade III gliomas, irrespective of relative astrocytic versus oligodendroglial histology.24 The possible consequences of this gene alteration are discussed later, but, notably, antibodies specific to the mutant form of the IDH1 protein can now be used reliably for glioma diagnosis on routine tissue sections.25 Interestingly, both TP53 and IDH1 mutations have been positively correlated with tumor MGMT methylation, which, in turn, is thought to accelerate G:C to A:T transition mutations. Others have previously noted that gliomas with MGMT methylation have a higher incidence of such mutations in TP53,26 and MGMT methylation has recently been correlated with IDH1 mutation in glioma,27 with the most common IDH1 alteration also being of the G:C to A:T transition type. Thus, it is possible that epigenetic alteration of low-grade glioma genomes, specifically methylation inhibition of MGMT expression, promotes the mutation of genes whose alteration are well established as occurring frequently in low-grade tumors.
Progression to Anaplastic Astrocytoma
The transition from WHO grade II astrocytoma to WHO grade III anaplastic astrocytoma is accompanied by a marked increase in malignant behavior.14 Although patients with grade II astrocytomas may survive for 5 or more years, patients with anaplastic astrocytomas often die within 2 or 3 years and frequently show progression to GBM. Histologically, the major differences between grade II and grade III tumors are increased cellularity and the presence of mitotic activity (Fig. 28.1B), implying that higher proliferative activity is the hallmark of the progression to anaplastic astrocytoma.
A number of molecular abnormalities have been associated with anaplastic astrocytoma, and several studies indicate that these abnormalities converge on one critical cell-cycle regulatory complex which includes the p16, cyclin-dependent kinase 4 (cdk4), cdk6, cyclin D1, and retinoblastoma (Rb) proteins. The simplest schema suggests that p16 inhibits the cdk6/cyclin D1 and/or cdk4/cyclin D1 complexes, preventing these from phosphorylating Rb, and so ensuring that phospho-Rb (pRb) maintains its brake on the cell cycle (Fig. 28.2).
Chromosome 9p loss occurs in approximately 50% of anaplastic astrocytomas and GBMs, and these 9p alterations target the CDKN2A locus, which encodes the p16 and ARF proteins. The CDKN2A gene is inactivated either by homozygous deletion or, less commonly by point mutations or hypermethylation.28,29 Loss of chromosome 13q occurs in one-third to one-half of high-grade astrocytomas, with the RB1 gene preferentially inactivated by losses and mutations. RB1 and CDKN2A alterations in primary gliomas are inversely correlated, and rarely occur together in the same tumor. Inactivation of pRb or p16 in mouse astrocytes has been shown to lead to anaplastic astrocytomas.30 Amplification of the CDK4 gene, located on chromosome 12q13-14, provides an alternative to subverting cell-cycle control and facilitating progression to GBM in up to 15% of malignant gliomas.28 Detection of any of the gene alterations known to influence Rb protein function (CDKN2A homozygous deletion, CDK4 amplification, or RB deletion + mutation) is associated with a poor prognosis for anaplastic astrocytoma patients.31
Allelic losses on chromosome 19q have been observed in up to 40% of anaplastic astrocytomas and GBMs, indicating a progression-associated glial tumor suppressor gene that maps to 19q13.3, but the gene(s) being targeted for inactivation has is yet to be identified.
Progression to Glioblastoma
GBM is the most malignant grade of astrocytoma, with survival times of substantially less than 2 years for most patients. Histologically, these tumors are characterized by dense cellularity, high proliferation indices, microvascular proliferation (Fig. 28.1C) and focal necrosis (Fig. 28.1D). The highly proliferative nature of these lesions is most likely the result of multiple mitogenic effects. As previously mentioned, at least one such effect is deregulation of the p16-cdk4/6-cyclin D1-pRb pathway of cell-cycle control (Fig. 28.2). The vast majority, if not all, GBM have alterations of this system, whether it involve inactivation of p16 or pRb, or overexpression of cdk4.9,28,29,32
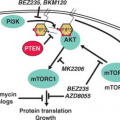
Chromosome 10 loss is a frequent finding in GBM, occurring in 60% to 95% of these tumors, and is far less commonly observed in anaplastic astrocytomas. The PTEN tumor suppressor gene at 10q23.3 is clearly one target of the chromosome 10 deletions, with PTEN mutations of the remaining allele identified in up to 30% of GBM, and a lesser percent of GBM having deletion of all or part of their remaining PTEN gene.21
Only gold members can continue reading. Log In or Register to continue