Fig. 1
Pathophysiology in the subentities and variants of MCL. MCL regroups at least three diseases: the classical nodal MCL, the most common entity, the indolent leukemic/splenic MCL, and the blastoid variant associated with a very aggressive behavior with poor response to treatment. The translocation t(11;14)(q13;q32) and the overexpression of cyclin (CCND1) is acquired in pre-B cells of the bone marrow and is followed by two different molecular pathways that configure the two clinical and biological subtypes of the disease: the nodal MCL and the leukemic/splenic MCL. The blastoid variant may be diagnosed either at the beginning of the evolution or later during the course of the disease. The outcome is very poor
A subset of MCL does not carry the t(11;14) translocation and CCND1 expression but they have the same pathological and clinical characteristics of MCL with this genetic alteration [15].
The most common alterations further deregulating cell cycle in MCL involve the INK4a/CDK4/RB1 and ARF/MDM2/TP53 pathways [13]. The relevance of cell cycle deregulation in MCL is highlighted by the poor prognosis conferred by high proliferative activity measured either by a gene expression signature or the Ki-67 index [16]. Recent genome-wide study using next-generation sequencing (NGS) has expanded the perspective of genes and pathways involved in the development of MCL. These studies have confirmed that the most common secondary alteration in MCL is the mutation of the DNA damage sensor ATM, usually associated with 11q deletions and a high number of chromosomal alterations [17]. These alterations seem to accumulate in tumors expressing SOX11 [17]. Novel mechanisms identified include activating mutations in NOTCH1/2 in around 10 % of the tumors associated with an aggressive evolution [18]. Mutations in several chromatin modifiers such as WHSK1, MLL2, and MEF2B have been also detected almost exclusively in MCL expressing SOX11 [17]. Somatic mutations in regulatory genes of the NFKB pathway have been identified in around 10–15 % of MCL [17]. BIRC3 is the most commonly affected gene (6–10 %). Other alterations in both canonical and alternative NFKB pathways include recurrent inactivating mutations of TRAF2, activating mutations of TLR2, and occasional mutations of CARD11, MAP3K1 (NIK), and IKBKB (IKKB), suggesting that NFKB pathway may be activated by genetic alterations in a higher number of cases that initially thought. A practical consequence of these alterations is their possible relationship with resistance of MCL cells to inhibitors of the BCR pathway as demonstrated in MCL cell lines [19].
In addition to frequent genetic alterations, MCL has deregulation of different signaling pathways that may be important targets of new drugs [13].
The promising results obtained with the inhibitors of the BCR signaling suggest that survival of MCL cells depends on the activation of this pathway, although the mechanisms are not entirely understood [20]. Activation of the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway has been observed in MCLs and is an effective target for new therapies [13]. Recent studies are emphasizing the potential relevance of the interactions between MCL cells and the microenvironment [21, 22].
3 Clinical Presentation
The clinical presentation of MCL is very different regarding the subtypes: nodal or leukemic/splenic. In the classical nodal MCL, representing 80 % of the patients, the disease is nodal and disseminated, including generalized lymphadenopathies and bone marrow involvement. The Ann Arbor is stage III or IV. Bulky disease and B symptoms are uncommon [23]. Extranodal involvement is almost constant, the most frequent sites involved being the bone marrow, blood, and colon. This intestinal involvement is usually asymptomatic. The clinical behavior of MCL patients is aggressive with a median overall survival (OS) of around 3–4 years 10 ago. However the OS has been dramatically improved with median at around 7 years in 2015 based on recent advances in therapeutics [24, 25].
Leukemic involvement may also appear during the evolution of the disease and could represent a manifestation of progression to blastoid variant. These cases are usually associated with a median survival of only 3 months [26].
The patients with an indolent leukemic/splenic MCL present with an indolent disease and no B symptoms. The clinical exam confirms the presence of a splenomegaly that may be massive and no lymphadenopathy. This small proportion of patients has longer survival, even without the need of any treatment.
4 Diagnosis
The diagnosis of MCL is established according to the criteria of the WHO classification of hematological neoplasms [1] based on a nodal biopsy. In the case of indolent leukemic/splenic MCL subtype, diagnosis is based on the immunophenotype of the peripheral blood B cells and bone marrow biopsy. Besides the classical immunophenotype (CD5+/CD19+/CD20+/CD22+ and CD10−/CD23−/CD200−), the detection of the pathognomonic cyclin D1 overexpression (immunohistochemistry) or the chromosomal translocation t(11;14) by fluorescent in situ hybridization is crucial, since histomorphological phenotypes may differ significantly. In addition, rare cases of cyclin D1-negative variant of MCL have been recognized, characterized by a similar gene expression profile and numerous secondary genomic alterations as classical MCL. SOX11, a transcription factor expressed in 90 % of MCL, might be helpful to identify the cyclin D1-negative variants.
Moreover, both the cell proliferation gene expression pattern and the Ki-67 proliferative index staining represent powerful prognostic indicators of long-term outcome.
The indolent leukemic/splenic MCLs also exhibit the chromosomal translocation t(11;14) and the overexpression of CCD1. They predominantly display hypermutated immunoglobulin genes, non-complex karyotypes, and a peculiar gene expression profile.
5 Prognostic Parameters
The different therapeutic approaches do not consider the high heterogeneity in the evolution of the disease in MCL patients. The increasing number of therapeutic options is opening new perspectives for patients but the evaluation of these approaches will require a correct stratification of the patients according to the specific biological risk of their disease. Recently, a specific MCL prognostic index (MIPI, Mantle Cell Lymphoma International Prognostic Index) based on four independent prognostic factors [age, Eastern Cooperative Oncology Group (ECOG) performance score, lactate dehydrogenase (LDH), and leukocyte count] has shown the capacity to clearly separate MCL patients into three groups with significantly different prognoses [27].
As described in the chapter describing the physiopathology of MCL, the proliferation of the tumor evaluated either as the mitotic index or cells expressing the proliferation-associated antigen Ki-67 is the best predictor of survival in MCL patients and has an independent prognostic value from the MIPI [28]. The analysis of gene expression profiling in MCL has identified a proliferation signature based on the expression of 20 genes able to clearly discriminate patients with different median survival, confirming that increased proliferation was the best predictor of poor survival [29]. New markers independent of the proliferation have been recently reported to influence the behavior of the disease or the response to the current treatment. Thus the concomitant inactivation of the two regulatory pathways, INK4a/CDK4 and ARF/p53 in MCL, was associated with a poor survival that was independent of Ki-67 proliferation index [30].
6 Treatment
In countries where treatment is available, the initial therapeutic decision for a patient with MCL is dictated by the age and, more importantly, the fitness of the patients (Fig. 2). For years the standard treatment was the classical CHOP (cyclophosphamide, Adriamycin, vincristine, and prednisone) for fit patients or chlorambucil for frail patients. The complete remission rate was 20–50 % and the median OS of around 3 years. The intensive leukemic regimen hyper-CVAD (a dose-intense, hyperfractionated cyclophosphamide in a CHOP-like combination plus high-dose methotrexate and cytarabine) improved the results of CHOP in non-randomized studies in younger patients [31]. Fludarabine-containing regimens and other polychemotherapy combinations were also used with no substantial modifications on the outcome of the patients. More recently, the strategies for the management of MCL patients have changed due to the introduction of immunotherapy and new drugs that are more targeted to molecular mechanisms of the disease. The combination of chemotherapy regimens such as CHOP, hyper-CVAD, or FCM (fludarabine, cyclophosphamide, mitoxantrone) with rituximab, a chimeric monoclonal anti-CD20 antibody with limited efficacy as a single agent, has been shown to produce impressive overall response rates of up to 80–95 % and CR rates of up to 30–87 % in previously untreated patients [32–34].
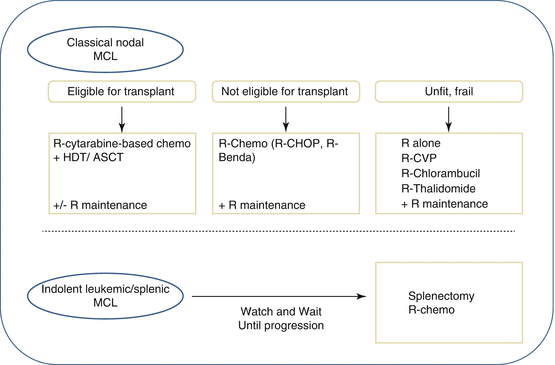
Fig. 2
Treatment strategies in MCL. The initial therapeutic decision for a patient with MCL is dictated by the age and, more importantly, the fitness of the patients. For the fit patients eligible for high-dose therapy and autologous stem cell transplantation (HDT/ASCT), the treatment of choice involves a cytarabine-based regimen (R-DHAP/X/C (rituximab, dexamethasone, high-dose aracytine + cisplatin, or oxaliplatin or carboplatin) or hyper-CVAD), which is consolidated with a high-dose therapy associated with an autologous transplant. Rituximab in maintenance has been recently proved to increase PFS. For patients in whom an intensive approach to management is not feasible, several possibilities of immunochemotherapy may be proposed to the patients such as R-CHOP and R-bendamustine. In this population, there is a clear benefit of rituximab as maintenance after R-CHOP. For frail and unfit patients, a number of less intensive therapies are available, including rituximab alone as well as CVP, cladribine, or thalidomide, usually in combination with rituximab. For patients with indolent leukemic/splenic MCL, watch and wait should be observed until progression. At time of treatment decision, several options may be proposed such as splenectomy and R-chemotherapy. HDT/ASCT high-dose therapy and autologous stem cell transplantation
These progressive therapeutic improvements are now the basis of the MCL treatment in first line. For the fit patients eligible for high-dose therapy and autologous stem cell transplantation (HDT/ASCT), the treatment of choice involves a cytarabine-based regimen (R-DHAP/X/C (rituximab, dexamethasone, high-dose aracytine + cisplatin, or oxaliplatin or carboplatin) or hyper-CVAD), which is consolidated with a high-dose therapy associated with an autologous transplant. This strategy produces impressive results with a CR/CRu rate before transplant at 77 % and after transplant 92 % (Cheson 1999 criteria) [35], further improved by a rituximab maintenance after transplant with a 2y-EFS of 93.2 % (95 % CI, 86.9–96.6) in the rituximab arm versus 81.5 % (95 % CI, 72.7–87.7) in the WW arm (HR = 2.1) [36]
For patients in whom an intensive approach to management is not feasible, several possibilities of immunochemotherapy may be proposed. The most commonly used are R-CHOP [33], R-fludarabine and cyclophosphamide (FC) [37], and R-bendamustine [38]. A large randomized study recently demonstrated a survival benefit for the use of R-CHOP over rituximab and FC (R-FC) in older patients with MCL [39]. This study also demonstrated the clear benefit of rituximab as maintenance after R-CHOP, where it doubled remission duration in responding patients. The use of the R-CHOP/R-cyclophosphamide, vincristine, and prednisolone (CVP) regimens in comparison with R-bendamustine (R-B) has been assessed across a range of lymphomas including MCL in two randomized trials [38, 40]. For the MCL cohorts, the R-B combination demonstrated a superior PFS [38] and response rate [40] but no difference in OS. Neither trial involved rituximab maintenance; because the addition of this significantly improves the outcome following R-CHOP and there are currently no available data on maintenance after R-B, it is not clear which of these two regimens is superior. Recently it has been shown in a randomized trial that the association of bortezomib to R-CHOP improves the response rate and the PFS compared to R-CHOP alone [41].
For the more frail patients where it is not possible to use either of these approaches, a number of less intensive therapies are available, including rituximab alone as well as CVP, chlorambucil, cladribine, or thalidomide, usually in combination with rituximab [25].
7 New Therapies
The growing insights into the molecular biology of MCL lead to the systematic exploration of targeted approaches. Interestingly, in contrary to the short-term remissions after chemotherapy, various molecular approaches achieve the highest response rates in this distinct lymphoma subtype. Thus, proteasome inhibitors (bortezomib), immune modulatory drugs (lenalidomide), or mTOR inhibitors (temsirolimus) have been meanwhile registered in relapsed MCL [25] for review. Inhibitors targeting BCR signaling pathway are highly active in mantle cell lymphoma. In an international phase II trial, the Bruton’s kinase (BTK) inhibitor ibrutinib achieved an overall response of 68 % (CR 21 %) [20]. Results of phase III trials are eagerly awaited comparing ibrutinib vs. temsirolimus in relapsed disease and assessing BR schedule +/− ibrutinib in first line. Another inhibitor of the BCR signal cascade, the delta-specific PI3K inhibitor (idelalisib) also achieved a promising ORR of 32 % in relapsed MCL, but duration of remission seems to be limited [42]. Two cell cycle targeted drugs, flavopiridol and PD0332991 (direct inhibitor of cyclin-dependent kinase 4 and 6), showed also activity in relapsed MCL alone and in combination with fludarabine, rituximab, or bortezomib. Finally, promising results have been recently reported for an oral second-generation BCL-2-specific MH3-mimetic ABT-199.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


