15 Cancers of the musculoskeletal system
Primary bone tumours
Aetiology
For osteosarcoma the most important risk factor is prior radiotherapy which accounts for only 3% of cases, with a time interval of 14 years (range 4–40 years). It is the most common secondary malignancy following treatment for childhood cancer. Other factors include chemotherapy with alkylating agents, Paget’s disease, chronic osteomyelitis, and hereditary conditions such as hereditary retinoblastoma and Li–Fraumeni syndrome (p. 53, 55). In hereditary retinoblastoma, the relative risk (RR) of osteosarcoma is 500 for limb osteosarcomas and 2000 for skull osteosarcomas following irradiation for retinoblastoma. Li–Fraumeni syndrome patients have a RR of 15 for osteosarcoma at all sites.
Pathogenesis
Ewing’s tumours consistently have reciprocal chromosomal translocations, usually between the EWS gene on chromosome 22 and FLI-1 on chromosome 11. In 85% of cases this is t(11;22)(q24;q12). Other variants place EWS with other genes such as t(21;22)(q22;q12) EWS-ERG translocation which occurs in up to 10%.
Clinical features
There are some features which are common to many types of bone tumour (Box 15.1).
Box 15.1
Clinical features seen in many types of bone tumour
In advanced disease systemic symptoms may be present. These include
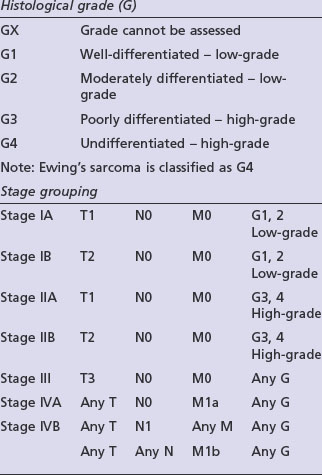
Staging
Adequate staging includes optimal imaging of the primary site (MRI) and the entire bone to exclude intra-medullary skip metastases, which occur in osteosarcomas and Ewing’s tumours. A CT scan of the chest should be done to assess for possible pulmonary metastases. For PNETs arising in the lower half of the body a CT of the pelvis and abdomen can be considered as there may be lymph node involvement. A bone scintigram, or in some centres a CT-PET, will assess for bone metastases. A whole body MRI on STIR sequence has shown to be effective in detection of bone metastases which may not be visible with other forms of imaging, particularly in Ewing’s tumours. A bone marrow aspirate should be performed in patients with a Ewing’s tumour to exclude bone marrow involvement. Staging is according to the AJCC bone tumour staging (Table 15.1).

Osteosarcoma
Several subtypes of osteosarcoma exist. Over 90% are high-grade intramedullary tumours, half of which are osteoblastic and the other half are split equally between fibroblastic and chondroblastic osteosarcomas. The remaining 10% are made up of small subgroups including telangiectatic osteosarcoma, extraosseous osteosarcoma, juxtacortical osteosarcoma and malignant fibrous histiocytoma of bone which is treated in the same way as intramedullary osteosarcoma.
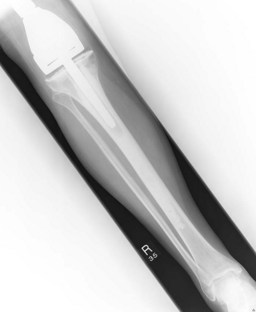
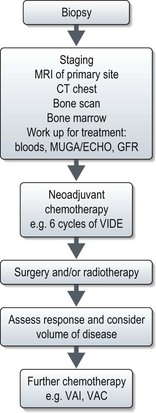
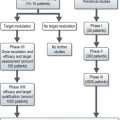
Figures 15.1 and 15.2 show imaging features of osteosarcoma. Box 15.2 shows prognostic factors in osteosarcoma. An overview of management is shown in Figure 15.3.
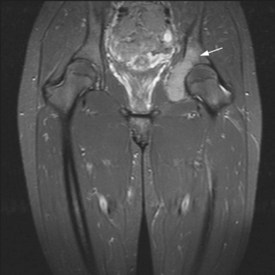
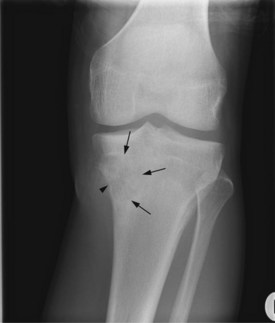
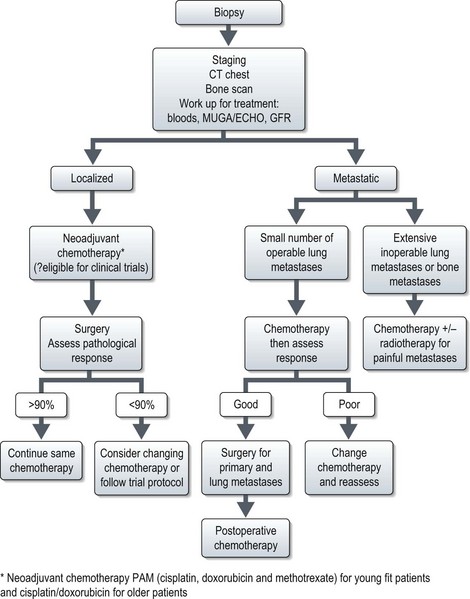
Localized osteosarcoma
Localized osteosarcoma is treated with neoadjuvant chemotherapy followed by surgery (Figure 15.4) and adjuvant chemotherapy.
Chemotherapy
Prior to the 1980s all patients with apparently localized disease had surgery (usually amputation) but only 20–30% survived over 5 years. Most died of disseminated disease suggesting the presence of micrometastases in apparently early disease. A pivotal study showed that giving adjuvant chemotherapy improved survival from 17% to 66%, and neoadjuvant chemotherapy was subsequently implemented. The most frequently used drugs are cisplatin and doxorubicin. Methotrexate at doses up to 12 g/m2 is used in addition to cisplatin and doxorubicin in younger patients and has been found to increase tumour necrosis. The degree of tumour necrosis after neoadjuvant chemotherapy is of prognostic value. Less than 90% necrosis is associated with a poorer prognosis. Whether prognosis can be improved by changing chemotherapy after surgery is one of the roles of the international EURAMOS trial.
Metastatic osteosarcoma
Chemotherapy
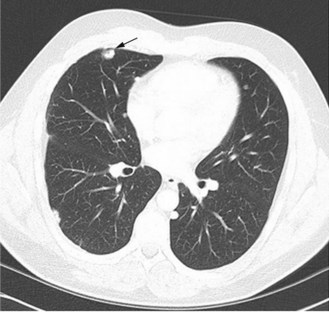
As in the adjuvant setting, the most effective agents are cisplatin, doxorubicin, methotrexate and ifosfamide (Box 15.3). Response rates are between 20–40% although some tumours and metastases may not reduce in size. Apparent calcification of lung metastases can occur (Figure 15.5). If the presentation is with metastases, combination chemotherapy should be the primary treatment.
Box 15.3
Chemotherapy for metastatic osteosarcoma
Ewing’s sarcoma
Presentation
Unlike osteosarcomas which present most often in the epiphyses of long bones, Ewing’s sarcomas occur within the diaphysis and are often associated with a soft tissue mass. The soft tissue reaction may be confused for infection and contribute to delay in diagnosis. The median delay from symptoms (Box 15.1) to diagnosis is 6–9 months. The most common sites are long bones (53%) or the axial skeleton (47%). 25% have a soft tissue primary. Systemic symptoms (fever, anorexia, weight loss, and lethargy) may be present and are associated with advanced disease. 25% patients have metastases at presentation. Subclinical metastases are present in 80–90% with apparently localized disease, necessitating multimodality treatment even in apparently small volume localized disease. Prognostic factors are listed in Box 15.4.
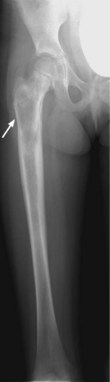
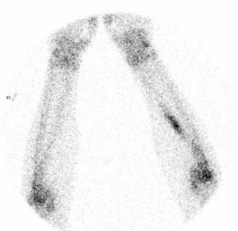
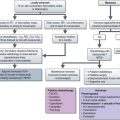
X-ray may show a characteristic destructive lesion within the bone with surrounding periosteal reaction known as an onion skin appearance (Figure 15.6). A soft tissue reaction may also be seen on X-ray and a pathological fracture is seen in 15%. Ewing’s sarcomas, like osteosarcomas, show intense tracer uptake in bone scan (Figure 15.7). Staging is given in Table 15.1 and Figure 15.8 shows an overview of management.
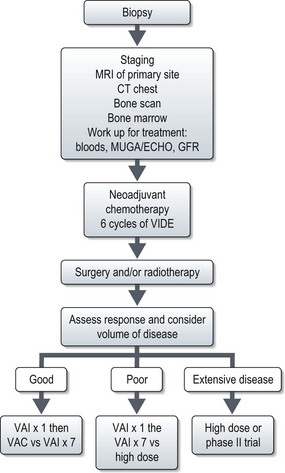
Management of localized disease
Chemotherapy
Stratifying treatment according to risk is under investigation in the EUROEWING99 trial (Figure 15.9). Low-risk patients are randomized between standard treatment and less intense treatment. High-risk/metastatic patients are randomized between standard treatment and high-dose with stem cell rescue.
Radiotherapy
Unlike osteosarcomas, Ewing’s tumours are radiosensitive and, radiotherapy used to be the principle method of achieving disease control. Studies have shown radiotherapy without surgery has a worse outcome than surgery for local control but there may be some selection bias. However, radiotherapy still remains a primary method of local control in unresectable tumours when doses of 45–55 Gy have been used (Box 15.5).
Box 15.5
Radiotherapy in sarcomas
Indications
Clinical target volume


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


