Cancers in Childhood
Theodore B. Moore
Carole G. H. Hurvitz
INCIDENCE AND SURVEILLANCE
I. INCIDENCE AND OVERVIEW.
Although cancer is the second leading cause of death in children (12% of deaths), it is still relatively uncommon. The incidence of cancer is increasing, however. Fortunately, with modern aggressive multidisciplinary therapy, 5-year survival rates for children with cancer exceed 75%.
A. Cooperative groups. The treatment of children with cancer is highly specialized. Whenever possible, patients younger than 18 to 21 years of age should be treated in specialized centers related to one of the major pediatric cooperative groups, such as the Children’s Oncology Group. More than 90% of children younger than 10 years of age are treated in such centers, and their mortality has decreased proportionally. Only about 30% of teenagers are enrolled in such centers, however, and the mortality rates in this group have not shown the same improvement.
B. Incidence. Leukemia and lymphoma make up almost half of the cases of malignancy in childhood, followed by central nervous system (CNS) tumors. The mortality rate for CNS cancers now exceeds that for acute lymphocytic leukemia.
There is no formal reporting system for malignant tumors in children in the United States. SEER (Surveillance, Epidemiology, and End Results) reports from the National Cancer Institute indicate that approximately 164 cases of cancer occur per 1 million population <20 years of age in the following incidences per million:
|
The COG has recently set up a registry to try and determine the incidence.
II. LONG-TERM SURVIVAL AND SURVEILLANCE.
Now that more or most children with cancer are cured, the complications of the disease and its management are becoming increasingly important. In addition, some of the long-term complications are just now being appreciated. The most significant complications are as follows:
A. Neurocognitive development. Radiation to the brain and central nervous systems leads to learning disabilities and school problems. Fortunately, RT has been almost eliminated from leukemia treatment plans. Chemotherapy, especially methotrexate, but other agents also, can lead to learning difficulties.
B. Growth retardation. Radiation to the spine or limbs causes reduced growth in the affected area. Steroids and brain irradiation lead to endocrine and growth problems.
C. Second malignances. Breast cancer is a very significant problem in girls who have had radiation to the chest, especially for Hodgkin lymphoma.
The incidence reaches almost 40% by age 40 years. Even the newer reduceddose regimens are still associated with an increased incidence of breast cancer. Radiation leads to second cancers in the radiated area. Brain tumors were frequently seen in leukemia patients after CNS radiation. Having one cancer may put patients at risk of a second cancer. Cancer survivors are at risk of developing colon and skin cancers, leukemia, and lymphoma.
The incidence reaches almost 40% by age 40 years. Even the newer reduceddose regimens are still associated with an increased incidence of breast cancer. Radiation leads to second cancers in the radiated area. Brain tumors were frequently seen in leukemia patients after CNS radiation. Having one cancer may put patients at risk of a second cancer. Cancer survivors are at risk of developing colon and skin cancers, leukemia, and lymphoma.
D. Fertility. High-dose chemotherapy can reduce oogenesis and spermatogenesis. Radiation to the pelvis, as in Wilms tumor, causes increased fetal losses due to damage to the uterus.
E. Cardiovascular and respiratory systems. Anthracyclines cause damage to the heart. Recent studies are showing that even lower doses, thought to be safe, can have long-term effects. The younger the child and the greater the dose, the greater the risk. Pregnant women who were treated for cancer as children may develop heart failure during delivery. Radiation to the lungs and drugs such as bleomycin and cyclophosphamide can damage the lungs and cause decreased respiratory reserve.
F. Recommended surveillance
1. Mammograms, or preferably breast MRI, and breast examination are recommended annually for girls with RT to the chest, especially for Hodgkin lymphoma. They should begin screening at 8 years after irradiation or age 25 years, whichever is first.
2. Echocardiograms are recommended every 2 to 5 years, depending upon the age at treatment and the dose. These recommendations are subject to change.
3. Colonoscopy is recommended beginning age 25 or 10 years after RT for those who have received radiation therapy to the pelvis and abdomen.
4. Skin examinations annually are recommended for patients following radiation and stem cell transplantation.
LEUKEMIA AND LYMPHOMA
I. ACUTE LEUKEMIA
(see Chapter 25)
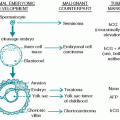
A. Pathology. Acute lymphoblastic leukemia (ALL) accounts for 80% to 85% of leukemias in childhood. Acute myelogenous leukemia (AML) accounts for 15% and chronic myelogenous leukemia accounts for 5% of cases.
In ALL, 15% to 25% of cases are T-cell, <5% are B-cell, and the remainder are precursor B-cell leukemias. Of the precursor B-cell leukemias, 70% possess the common acute lymphoblastic leukemia antigen (CALLA, CD-10). They are usually also terminal deoxynucleotidyl transferase-positive. Almost all are also CD-19-positive.
B. Treatment of ALL in childhood involves induction of remission, prophylaxis to the CNS, a phase of consolidation and reinduction, and maintenance therapy. Standard treatment for ALL leads to long-term remission in >85% of cases. Induction therapy employs vincristine, prednisone, dexamethasone, and L-asparaginase with the addition of daunomycin or doxorubicin, depending on risk stratification. Intensification therapy includes CNS prophylaxis. During maintenance therapy, oral mercaptopurine is given daily and methotrexate weekly for 2 to 3 years. Many patients receive monthly pulses of vincristine plus prednisone or dexamethasone. One or two cycles of a reinduction regimen are often added in ALL.
Certain prognostic factors at diagnosis affect the outlook of children with ALL, and their treatment is modified accordingly. Children with
poorer prognostic features require more intensive treatment than standard therapy.
poorer prognostic features require more intensive treatment than standard therapy.
1. Favorable prognostic factors for ALL. Average risk factors include initial white blood cell (WBC) count of <50,000/µL and age 1 to 9 years. Favorable features include pre-B subtype, L1 morphology, hyperploidy, lack of organomegaly, low bone marrow blasts on day 7 of induction therapy, trisomy of chromosomes 4 and 10, and t(4;11) or Tel/AML1 translocations. It has been shown that lack of minimal residual disease (MRD) at the end of induction gives a better prognosis or at least a positive MRD carries a much poorer prognosis.
2. Poor prognostic factors include WBC >50,000/µL, age <1 year or >10 years, massive organomegaly, lymphoma-like features, CNS involvement at diagnosis, mediastinal mass, failure to achieve remission by day 14 or 28, and certain chromosomal translocations, especially MLL gene rearrangements (11q23) in infants, the presence of the Philadelphia chromosome, and positive MRD at the end of induction or at any other time.
3. AML (Acute Myeloid Leukemia) accounts for 15% to 25% of leukemias in childhood.
In AML, high-risk features include monosomy 7, monosomy 5, 5q deletions, and FLT3 mutations. FLT3/ITD (internal tandem duplication) is a poor prognostic factor. In adults, 20% to 30% are FLT3 positive. Secondary AML is particularly difficult to treat and carries a poor prognosis. In children, only 5% to 17% are positive. Positivity increases with age. Good risk features include inv (16)/t(16;16), t(15;17), and t(8;21) conferring survival exceeding 70%.
Treatment of AML requires intensive chemotherapy. At the present time, hematopoietic stem cell transplantation (HSCT) is recommended for high-risk patients, preferably matched family donor (MFD) if available. MFD is recommended for intermediate risk if available and chemotherapy alone for low-risk patients.
C. Survival. The 5-year survival rate is >85% in children with “good-prognosis” ALL following standard therapy. Even children with poorer risk factors who receive intensive therapy have an overall long-term survival of at least 70%. Sites of relapse include the CNS, testes, and bone marrow. The risk for relapse after 2 years off therapy is very low.
The 5-year survival rate with the best available regimens for children with AML is 65% to 70% in first remission with favorable prognostic factors or when consolidated with a sibling donor HSCT and about 50% for those without. HSCT (allogenic, autologous, or matched unrelated) is also often recommended for patients with ALL and AML who relapse.
II. LYMPHOMA
A. Non-Hodgkin lymphoma (see Chapter 21). In pediatrics, lymphomas can be considered to be lymphoblastic or nonlymphoblastic and localized or nonlocalized. Lymphoblastic lymphomas are usually T cell and, when nonlocalized, may be the same entity as T-cell leukemia; these illnesses are usually treated in the same way. Nonlymphoblastic lymphomas are usually B cell and frequently were previously called Burkitts (or Burkitts-like) lymphoma.
Different combination chemotherapeutic regimens are necessary for the subtypes of lymphoma. Localized lymphomas respond very well to chemotherapy even when bulky, and have a cure rate of >90%. The prognosis for disseminated T-cell lymphomas is the same as for T-cell ALL. The outlook for disseminated nonlymphoblastic or B-cell lymphoma is about 50%.
B. Hodgkin lymphoma (see Chapter 21). There is no consensus on the treatment of Hodgkin lymphoma in children. Chemotherapy is used for all stages of disease. Staging laparotomy is no longer recommended. Splenectomy is contraindicated in young children because of fatal infectious complications and increased risk for leukemia. The alternation of the COPP and ABVD regimens (defined in Appendix D1) or a hybrid of them is frequently recommended rather than either regimen alone. In children, local-field rather than extended-field radiation is preferred in an effort to reduce long-term side effects, such as growth retardation and second cancers, especially breast cancer in girls.
Second malignancies are a major problem with the risk approaching 40% by age 35 years for girls who have been irradiated. Current Children’s Oncology Group (COG) treatment regimens are evaluating modulation of therapy based on initial response with a goal of minimizing toxicity while maintaining high cure rates.
BRAIN TUMORS
Neurologic malignancies are discussed in Chapter 14.
I. EPIDEMIOLOGY.
Brain tumors in children may be associated with certain underlying diseases including neurofibromatosis, tuberous sclerosis, and von Hippel-Lindau angiomatosis. Family clusters of CNS tumors have occasionally been reported.
II. PATHOLOGY AND NATURAL HISTORY
A. Pathology. Most CNS neoplasms in children are primary tumors of the brain; the single exception is meningeal metastases, which are common with leukemia and lymphoma. Astrocytomas are the most frequent type (about 50% of all cases). Medulloblastomas account for 25% of cases; ependymomas, 9% and glioblastomas, 9%.
B. Sites of disease. Brain tumors in children tend to occur along the central neural axis (i.e., near the third or fourth ventricle or along the brain stem). Most brain tumors that occur during the first year of life are supratentorial. In patients between 2 and 12 years of age, 85% are infratentorial. In patients >12 years of age, the relative incidence of supratentorial tumors increases.
III. SYMPTOMS AND SIGNS
A. Symptoms. The most common symptoms include headaches, irritability, vomiting, and gait abnormalities. Morning headaches are most characteristic, but drowsiness and abnormal behavior are also common. Symptoms may be intermittent, particularly in very young children who have open fontanelles. A head tilt is a common finding and often missed.
B. Physical findings include enlarged or bulging fontanelles in very young children and cerebellar abnormalities, papilledema, and sixth cranial nerve abnormalities in older children.
IV. TREATMENT AND SURVIVAL.
Survival rates for patients with low-grade astrocytomas are high if the tumor can be surgically removed (>90% at 5 years) and low if the tumor is high grade (<10% at 5 years). Survival for medulloblastoma depends on both local recurrence (<25% with surgery and radiotherapy) and spinal metastases (about 35% incidence without prophylactic spinal irradiation); this tumor is invariably recurrent when treated with surgery alone, but average risk patients have approximately 80% survival when treated with surgery, radiation, and chemotherapy.
Chemotherapy is now being used more frequently in children with brain tumors in an attempt to improve survival and to reduce the use of radiation, which has devastating effects in young children. RT is deferred in children <3 years of age and preferably in children <10 years whenever possible. Unlike childhood leukemia, relatively little improvement in survival has been obtained over the years. High-dose therapy with autologous HSCT support has shown promising results in certain disease types. In addition, experimental approaches using targeted therapies, novel chemotherapy and radiation delivery systems, and dendritic cell-based vaccines are currently under investigation.
NEUROBLASTOMA
I. EPIDEMIOLOGY AND ETIOLOGY.
Neuroblastoma is the most common congenital tumor and the most common tumor to occur during the first year of life. It rarely occurs in patients >14 years of age. About 40% occur in the first year of life, 35% from 1 to 2 years of age, and 25% after 2 years of age. Rarely, family clusters are reported.
II. PATHOLOGY AND NATURAL HISTORY.
Neuroblastoma has the highest incidence of spontaneous regression of any tumor in humans.
A. Histology. Neuroblastoma closely resembles embryonic sympathetic ganglia. The tumors partially differentiate into rosettes or pseudorosettes, mature ganglion cells, or immature chromaffin cells. Although histologically similar to ganglioneuromas and pheochromocytomas, neuroblastomas are clearly distinctive. Electron microscopy shows typical dendritic processes that contain granules with dense bodies, probably representing cytoplasmic catecholamines. The most primitive histologic type of neuroblastoma is composed of small round cells with scant cytoplasm. The ganglioneuroma is composed of larger, more mature ganglion cells with more abundant cytoplasm.
Homogeneously staining regions and double minute chromosomes seen in poor-prognosis neuroblastomas represent amplified N-myc segments. Amplification of N-myc is an intrinsic property of poor-prognosis tumors and can be rapidly detected by fluorescent in situ hybridization (FISH) concordant with Southern blot analysis.
B. Sites.




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
