Figure 20-1 Genomic alterations in glioma co-segregate with only some of the identified molecular subtypes. (With permission from Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98-110).
Recently, alternative approaches to those pursued using GWAS statistical approaches have started to emerge. 12 The rationale for these methods is that genome-wide regulatory models representing causal molecular interactions in the cell—for example, transcription factors regulating their transcriptional targets or protein kinases activating their substrates—may help us identify a relatively small number of candidate genes, upstream of genetic programs that are dysregulated, which may be tested for genetic and epigenetic alterations (Figure 20-3 ).
Variants of such a genetic genomics approach were pioneered in plants 13 and metabolic disease 14 and have been used successfully in cancer-related studies. For instance, identification of the novel HUWE1-MYCN-DLL3 cascade in brain tumors was possible by reverse engineering posttranslational modulators of MYCN activity as well as its downstream targets using reverse engineering algorithms. 15 Similarly, the role of RUNX1 as a tumor suppressor mutated in T-cell acute lymphoblastic leukemia (T-ALL) was elucidated based on its most significant overlap with the TLX1 and TLX3 oncogene regulatory programs. 16 In some cases, a network-based view of cancer biology may allow elucidation of the dependency of a phenotype on an entire collection of genetic events, which would be virtually impossible to dissect using statistical approaches. For example, it was recently shown that deletion of any combination of 13 genetic loci distributed across the entire genome leads to functional inactivation of PTEN in glioma patients, via a novel interaction mechanism involving competitive endogenous RNA (ceRNA). 17 Indeed, cancer systems biology applications have exploded over the past 3 years, with studies ranging from the study of key drivers of tumorigenesis in melanoma 18,19 to the dissection of tyrosine kinase signals downstream of ERBB receptors. 20,21
Such a regulatory-model–driven view of cancer biology is thus emerging as an important systems-level contribution to the study of this disease. By taking a more holistic view of tumor-related processes, anchored in gene regulatory mechanisms, cancer systems biology mediates the genetic and the genomic views of cancer to provide novel insight into its mechanisms. Specifically, the proponents of these approaches argue that among all genetic and epigenetic alterations in a tumor, those contributing to its initiation, progression, or drug sensitivity cannot affect regulatory interactions in a random way but must co-segregate within specific regulatory subnetworks that are thus globally dysregulated across different samples of a given tumor subtype. Hence, if the full complement of regulatory interactions regulating the behavior of a specific cancer cell population were known, then it should be able to use its structure to separate driver from passenger alterations. The example of RUNX1 in T-ALL 16 is particularly revealing in this case. Here, the functional role of RUNX1 mutations could only be elucidated after determining that its targets are virtually overlapping with those of two previously established oncogenes, TLX1 and TLX3. 22 Without this regulatory insight, it would have been impossible to identify these mutations as statistically significant across the full repertoire of genes.
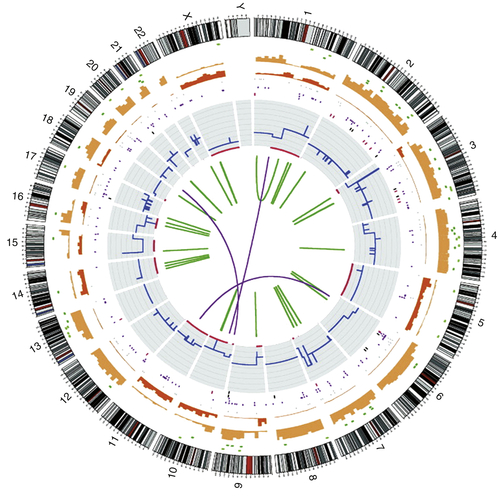
Figure 20-2 Circos plot showing the whole-genome catalogue of somatic mutations from the malignant melanoma cell line COLO-829 This genome has approximately 30,000 somatic base substitutions and 1000 somatic insertions and/or deletions. In coding exons, 272 somatic substitutions are present, including 155 missense changes, 16 nonsense changes, and 101 silent changes. The numbers and types of mutations are highly variable across different cancer genomes. Chromosome number and karyotype are indicated on the exterior of the plot. Key: blue lines, copy number across each chromosome; red lines, sites of loss of heterozygosity (LOH); green lines, intrachromosomal rearrangements; purple lines, interchromosomal rearrangements; red spots, nonsense mutations; green spots, missense mutations; black spots, silent mutations; brown spots, intronic and intergenic mutations (merged). (With permission from Garnett M, McDermott U. Exploiting genetic complexity in cancer to improve therapeutic strategies. Drug Discov Today. 2012;17:188-193).
A key issue, then, is how to assemble accurate and comprehensive repertoires of molecular interactions to create a quantitative regulatory model that may be interrogated to elucidate drivers of tumor-related phenotypes. This is an important question, because virtually all cancer-related publications today contain appealing graphical presentations of molecular pathways in cancer. These bona fide models could provide a starting point for a systems-level study of cancer, as proposed, for instance, by pathway-wide association study (PWAS) strategies. 23
Unfortunately, knowledge of molecular pathways governing physiological and tumor-related traits is still very poor. Indeed, canonical cancer pathways are more reflective of the researcher’s desire to understand biological processes as a relatively linear and interpretable set of events than of the true complexity of cellular regulation. Specifically, these representations have two major limitations. First, they are not context specific. For instance, the EGFR pathway would be identically represented for a glioma and for a lung-cancer cell.
Second, they constitute a manually curated collection of published facts, of which several are actually incorrect, and which represents less than 1% of the total complement of regulatory interactions in the cell. Hence, their use introduces a strong bias toward what is already known (prior knowledge). Indeed, in the absence of a prior hypothesis, interrogation of canonical cancer pathways has been largely unsuccessful in the elucidation of novel tumor-related mechanisms. To understand the difference between a true regulatory network and a canonical cancer pathway, consider Figure 20-4 , A, showing the differential phosphorylation of canonical EGFR pathway proteins in the H1650 cell line, where EGFR has an activating mutation, compared to the average of all cell lines. In contrast, Figure 20-4, B, shows the differentially phosphorylated proteins for the same cell lines in a signal transduction network, inferred de novo from a large-scale collection of phosphopeptide profiles of non–small-cell lung adenocarcinoma. 24 Whereas the pathway-based representation provides no clue that the EGFR pathway may be dysregulated, the network-based representation shows a clear hyperphosphorylated protein pattern surrounding both EGFR and MET.
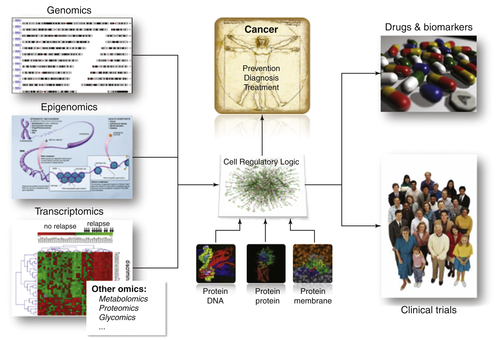
Figure 20-3 The -omics layers of the cell both encode and are processed by a context-specific regulatory logic. At the atomic level, this logic is implemented via molecular interactions, such as protein-DNA, protein-protein, protein-RNA, and RNA-RNA. Dissection and interrogation of this logic in context-specific fashion, using systems biology approaches, is starting to allow elucidation of driver genes responsible for the presentation of relevant cancer-related phenotypes.
In the following, we discuss the idea of a simultaneous, de novo reconstruction of context-specific gene regulatory networks from large-scale molecular profile data, and of the genetic and epigenetic variability they harbor and mediate. A classic systems biology workflow generally involves three steps: First is acquisition of molecular profiles for a variety of molecular species, several of which represent gene products, from mRNA to phosphopeptide abundance, as well as of genetic and epigenetic alterations. Second is data integration and reconstruction of the regulatory models for the specific cellular context of interest. The final step is regulatory model interrogation, using genetic and genomic signatures that represent the cellular states of interest. Given the abundance and prior coverage of molecular profile data for cancer, we concentrate on the two latter steps.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


