Cancer Stem Cells
Jean C. Y. Wang
John E. Dick
A fundamental problem in cancer research is identification of the cell type capable of initiating and sustaining growth of the tumor—the cancerinitiating cell or cancer stem cell (CSC). Although it has long been known that only a fraction of cells within a tumor is capable of tumor generation upon transplantation, it has been unclear until recently whether this observed functional heterogeneity was attributable to stochastic influences or to intrinsic properties of the tumor cells. Evidence for the existence of biologically distinct CSCs, first demonstrated in a hematological malignancy and in the past 5 years in several solid tumors, has shaped a new paradigm of human cancer as a hierarchical disease whose growth is sustained by a population of CSCs. This conceptual shift has important implications not only for researchers seeking to understand mechanisms of tumor initiation and progression, but also for the development and evaluation of effective anticancer therapies.
TUMOR HETEROGENEITY
Our modern understanding of the origin of cancer can be traced to Rudolph Virchow, who in 1858 stated the heretical and revolutionary thesis “omnis cellula e cellula,” that all cells come from cells. Out of his early theories grew the idea that cancer cells develop from normal cells that have undergone abnormal changes, a process now called somatic mutation. Nearly 150 years later, it is now well accepted that cancer is a genetic disease that arises from the clonal expansion of a single neoplastic cell. Simplistically, cancer has been viewed as the unregulated growth of abnormal cells, with the implication that all of the cells in the tumor are proliferating uncontrollably. However, this notion is incompatible with the observation made over four decades ago that only a fraction of cells within murine lymphomas were capable of clonogenic growth when transplanted into syngeneic mice.1 Furthermore, autotransplants of cancer cell suspensions in humans demonstrated that tumor growth occurred only after inoculation of at least 104 to 105 cells.2 These and similar observations demonstrated that there is functional heterogeneity in the proliferative ability of cells within a tumor.
Two contrasting theories have been proposed to explain this observed heterogeneity (Fig. 11.1).3 One view is that extrinsic factors (e.g., host resistance, growth factor concentrations, niche availability) or intrinsic factors (e.g., timing of cell cycle entry) prevent every cell from behaving in the same way. In other words, the behavior of tumor cells is unpredictable and governed by probabilities that may be influenced by any or all of these factors. The end result is that cells will appear to be heterogeneous in their proliferative capacity when tested in a functional assay. The central tenet of this stochastic model is that every cell has equal potential to initiate and sustain tumor growth, but most cells do not proliferate extensively due to the low cumulative probability of permissive events.
The alternative model is based on the biology of normal somatic tissues, many of which are arranged as hierarchies comprising cell types with different growth properties. The nonproliferating mature cells that make up the majority of these normal tissues must be continuously replenished by a pool of rapidly proliferating progenitors, which in turn are replenished by rare stem cells at the apex of the hierarchy. A key property of stem cells that distinguishes them from progenitors and allows them to maintain tissue integrity is self-renewal, whereby at cell division one or both daughter cells retain the biological properties of the parent cell. According to this model, tumors retain features of normal tissue organization, in that they are made up of distinct classes of cells that are organized hierarchically and possess intrinsically different functional capacities. The tumor hierarchy is sustained at its apex by a population of CSCs that possess the capacity to self-renew (i.e., produce more CSCs) and to recapitulate tumor heterogeneity by generating all of the various nontumorigenic cell types that compose the bulk of tumor. The essential principle of this model is that CSCs are biologically distinct from the bulk cell population, which does not possess tumor-initiating activity.
Both the stochastic and CSC models predict that only a small number of cells within a tumor will have the capacity to initiate tumor growth (i.e., to behave as CSCs); however, their underlying biological principles are very different. According to the stochastic model, the oncogenic program is operative in all cells of the tumor, thus
both research to understand neoplastic processes and drug development can be directed at the bulk cell population. In contrast, the CSC model implies that CSCs are biologically distinct from the majority of cells in the tumor due to irreversible epigenetic processes that influence cell function, layered onto the common genetic aberrations present in all cells of the tumor.4 The biological consequence of a particular cancer pathway may be different in CSCs compared to cells without tumor-initiating capacity. Thus, research must be directed at the relevant cell populations as identified through functional assays, the ultimate goal being the rational development of therapies that interfere with the oncogenic program within CSCs.
both research to understand neoplastic processes and drug development can be directed at the bulk cell population. In contrast, the CSC model implies that CSCs are biologically distinct from the majority of cells in the tumor due to irreversible epigenetic processes that influence cell function, layered onto the common genetic aberrations present in all cells of the tumor.4 The biological consequence of a particular cancer pathway may be different in CSCs compared to cells without tumor-initiating capacity. Thus, research must be directed at the relevant cell populations as identified through functional assays, the ultimate goal being the rational development of therapies that interfere with the oncogenic program within CSCs.
In order to test these theories and address the fundamental question of how tumor heterogeneity arises, two things are required: first, the ability to purify subpopulations of tumor cells based on physical or functional properties such as surface antigen expression or dye exclusion, and second, a functional transplantation assay to test the ability of purified cell populations to generate tumors in vivo, as current in vitro culture techniques do not reproduce the necessary microenvironment for tumor development. According to the stochastic model, the behavior of tumor cells is random and cannot be predicted; therefore, tumor-initiating activity will appear in every isolated cell fraction and cannot consistently be enriched. In contrast, the CSC model postulates that with an appropriate purification strategy, the CSCs with the capacity to initiate and sustain tumor growth in vivo can be identified and isolated from the bulk cells that do not have tumor-initiating activity (Fig. 11.1).
LEUKEMIA STEM CELLS
Based on the depth of knowledge gained from more than four decades of research in normal hematopoiesis, it is not surprising that identification of CSCs was first achieved in a hematological malignancy, acute myeloid leukemia (AML). This was made possible by prior detailed characterization of hematopoietic cell surface antigens and by the development of a xenotransplantation assay using severe combined immune-deficient (SCID) or nonobese diabetic (NOD)/SCID mice as recipients that allowed assessment of the ability of leukemic cells to initiate disease in vivo. When primary AML cells were fractionated based on expression of cell surface markers and transplanted into mice, only cells in the CD34+CD38− fraction composing less than 1% of the total blast population were able to initiate leukemic growth in vivo.5,6 These leukemia stem cells (LSCs) possessed high self-renewal, as demonstrated by serial transplantation, and proliferated in the mice to produce large numbers of leukemic progenitors and nonproliferating blasts, generating a graft that recapitulated the phenotypic and functional heterogeneity of the patient’s disease. These findings demonstrated that AML, like the normal hematopoietic system, is organized as a hierarchy of functionally distinct classes of cells whose growth is sustained by a small number of LSCs, which provides the first direct evidence supporting the CSC model.
LSCs share some phenotypic characteristics with normal hematopoietic stem cells (HSCs), for example expression patterns of CD34 and CD38. However, identification of LSC-specific markers such as the interleukin-3 (IL-3) receptor alpha chain (IL-3Rα, CD123)7 has allowed researchers to distinguish LSCs from normal HSCs. Such markers not only provide a therapeutic window for monoclonal antibody-based therapies to selectively target LSCs while sparing normal HSCs,8 but also enable elucidation of the unique properties of LSCs. For example, recent studies have demonstrated constitutive activation of the transcription factor nuclear factor κB (NF-κB) in quiescent primitive CD34+CD38−CD123+ AML cells but not in normal CD34+CD38− cells.9 Treatment of primitive AML cells in vitro with the NF-κB inhibitor parthenolide resulted in rapid induction of cell death and loss of ability to generate a leukemic graft in NOD/SCID mice, whereas normal CD34+CD38− cells were generally unaffected,10 suggesting that NF-κB plays an important role in the survival of LSCs but not normal HSCs. PTEN is a phosphatase that negatively regulates cell proliferation and survival through the phosphatidylinositol 3-kinase (PI3K) pathway. This pathway has been implicated in the survival of human AML LSCs,11 a finding supported by evidence that loss of PTEN in murine HSCs results in leukemia.12,13 Notably, treatment of PTEN-deficient mice with rapamycin, which targets the PI3K effector mammalian target of rapamycin (mTOR), prevented leukemia development and restored normal function to HSCs.12 Both NF-κB and PTEN thus represent exciting potential targets in the development of therapeutic strategies to kill AML LSCs while sparing normal HSCs.
Normal HSCs require a microenvironmental niche for maintenance of stem cell properties. If AML LSCs possess unique requirements for interaction with a supportive niche, this association could represent another therapeutic target. CD44 is a ubiquitously expressed transmembrane protein that mediates cell adhesion. Some isoforms of CD44 are highly expressed on AML blasts and are associated with poor prognosis. Treatment of AML cells in vitro or in vivo with an activating monoclonal antibody (H90) directed against CD44 resulted in killing of LSCs, as demonstrated by loss of leukemic repopulation in NOD/SCID mice, while similarly treated normal cord blood and bone marrow cells were much less affected, if at all.14 The mechanisms underlying eradication of LSCs included interference with homing to their microenvironmental niche, loss of engraftment ability, and induction of differentiation. These findings demonstrate that the leukemogenic process does not abrogate the niche dependence of
LSCs and highlight a potential therapeutic target that may also be applicable to solid cancers.
LSCs and highlight a potential therapeutic target that may also be applicable to solid cancers.
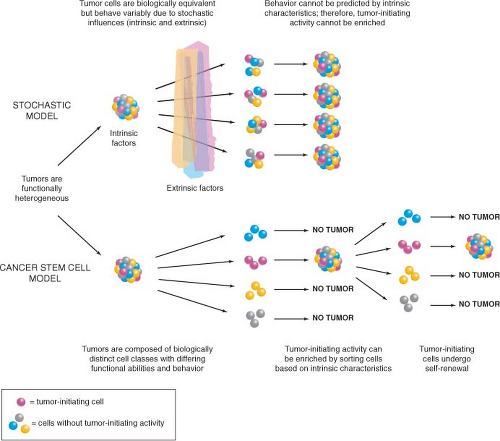 FIGURE 11.1 Models of tumor heterogeneity. Tumors are composed of phenotypically and functionally heterogeneous cells. There are two theories as to how this heterogeneity arises. According to the stochastic model, tumor cells are biologically equivalent but their behavior is influenced by intrinsic and extrinsic factors and is therefore both variable and unpredictable. Thus, tumor-initiating activity cannot be enriched by sorting cells based on intrinsic characteristics. In contrast, the cancer stem cell model postulates the existence of biologically distinct classes of cells with differing functional abilities and behavior. Only a subset of cells has the ability to initiate tumor growth; these cancer stem cells possess self-renewal and give rise to nontumorigenic progeny that make up the bulk of the tumor. This model predicts that tumor-initiating cells can be identified and purified from the bulk non-tumorigenic population based on intrinsic characteristics. (Figure originally published in Dick JE. Stem cell concepts renew cancer research. Blood 2008;112:4793.) © The American Society of Hematology. |
CANCER STEM CELLS IN SOLID TUMORS
Breast Cancer Stem Cells
Investigations of mechanisms underlying solid tumor heterogeneity were first undertaken in human breast cancer. Al Hajj et al.15 made single-cell suspensions of breast cancer specimens obtained from primary or metastatic sites in patients. Upon injection into the mammary fat pad of immune-deficient NOD/SCID mice, all samples studied were able to generate tumors. Thus, the NOD/SCID model provides a functional assay of the in vivo tumor-initiating ability of human breast cancer cells. Breast cancer cells are heterogeneous with respect to expression of a variety of surface markers, including the adhesion molecules CD24 and CD44. To test whether it would be possible to identify and isolate subpopulations enriched for tumor-initiating activity, breast cancer cells were first separated from normal hematopoietic, endothelial, mesothelial, and fibroblast cells by elimination of cells expressing lineage markers, then subfractionated based on
expression of CD24 and CD44. All of the in vivo tumor-initiating activity was found in the CD44+CD24−/lowLineage− cell fraction, with enrichment compared to unfractionated tumor cells as judged by the cell dose required for tumor formation. CD44− and CD44+CD24+Lineage− cells, even though morphologically indistinguishable from tumor-initiating cells, did not generate tumors at injection sites. In some samples, isolation of cells expressing epithelial specific antigen (ESA) allowed further enrichment of tumor-initiating activity within the CD44+CD24−/lowLineage− population, however, ESA expression did not distinguish between tumorigenic and nontumorigenic cells in at least one sample, and therefore may not be a reliable marker for breast cancerinitiating cells. CD44+CD24−/lowLineage− tumorigenic cells could be serially propagated, demonstrating self-renewal, and gave rise not only to more CD44+CD24−/lowLineage− tumorigenic cells, but also to the phenotypically diverse nontumorigenic cells which made up the bulk of the primary tumor, thereby recapitulating the tumor’s complexity and functional heterogeneity. This study was the first to isolate tumor-initiating cells from the bulk nontumorigenic population in a nonhematological malignancy, providing strong evidence that the growth of at least some types of human solid tumors is sustained by biologically distinct CSCs.
expression of CD24 and CD44. All of the in vivo tumor-initiating activity was found in the CD44+CD24−/lowLineage− cell fraction, with enrichment compared to unfractionated tumor cells as judged by the cell dose required for tumor formation. CD44− and CD44+CD24+Lineage− cells, even though morphologically indistinguishable from tumor-initiating cells, did not generate tumors at injection sites. In some samples, isolation of cells expressing epithelial specific antigen (ESA) allowed further enrichment of tumor-initiating activity within the CD44+CD24−/lowLineage− population, however, ESA expression did not distinguish between tumorigenic and nontumorigenic cells in at least one sample, and therefore may not be a reliable marker for breast cancerinitiating cells. CD44+CD24−/lowLineage− tumorigenic cells could be serially propagated, demonstrating self-renewal, and gave rise not only to more CD44+CD24−/lowLineage− tumorigenic cells, but also to the phenotypically diverse nontumorigenic cells which made up the bulk of the primary tumor, thereby recapitulating the tumor’s complexity and functional heterogeneity. This study was the first to isolate tumor-initiating cells from the bulk nontumorigenic population in a nonhematological malignancy, providing strong evidence that the growth of at least some types of human solid tumors is sustained by biologically distinct CSCs.
Other investigators have since shown that breast CSCs can be isolated from some patient tumors by cellular expression of aldehyde dehydrogenase (ALDH), with partial overlap with the CD44+CD24−Lineage− phenotype.16 As few as 20 ALDH1+CD44+CD24−Lineage− cells from one patient cancer could generate tumors in NOD/SCID mice. Although highly enriched, these CSC-containing fractions are still heterogeneous, and additional CSC markers will be required for further purification. Nevertheless, identification of breast CSCs has moved cancer researchers away from studying bulk tumors and shifted their focus to understanding the biology of this subpopulation of cells. For example, Yu et al.17 have shown that cell fractions enriched for breast CSCs have globally reduced microRNA expression compared to more differentiated cancer cells. In particular, the let-7 family is not expressed and increases with differentiation. Lentiviral expression of let-7 in Lineage−CD44+CD24− cells from patient breast cancers significantly reduced tumor formation in both primary and serially transplanted NOD/SCID mice. Insight into the biology of CSCs will be a crucial first step to develop an effective means to target them therapeutically.
Brain Cancer Stem Cells
Studies in several types of human brain cancers have clearly shown that the tumor cell population is functionally heterogeneous, in that only a fraction of cells have the ability to form tumor neurospheres when plated at low density in culture or generate tumors when transplanted in vivo.18,19 As discussed above, demonstration that this heterogeneity arises from the existence of biologically distinct cell populations, rather than as a result of stochastic processes, requires isolation of tumor-initiating cells from the bulk nontumorigenic population. Singh et al.20 reported that CD133+ cells in different types of human brain tumors possess extensive proliferative, differentiative, and self-renewal capacity in vitro. The development of a xenograft assay that involved injection of single-cell suspensions of human brain tumor samples into the NOD/SCID mouse brain enabled assessment of whether the CD133+ cells were capable of initiating tumor growth in vivo.21 Tumors could be generated by as few as 100 CD133+ cells, while injection of up to 105 CD133− tumor cells did not result in tumor formation. Importantly, small numbers of viable CD133− tumor cells could be found at the injection site many weeks later, ruling out the possibility that CD133− cells did not form tumors simply because they died following transplantation. The tumors generated by CD133+ cells resembled the patient’s original tumor by immunohistochemistry and consisted of a minority CD133+ and a majority CD133− cell population. CD133+ cells isolated from xenograft tumors could generate phenotypically similar tumors in secondary mice. Thus, CD133+ cells from human brain tumors possess the two key properties of CSCs: the ability to self-renew and to recapitulate tumor heterogeneity through differentiation. Interestingly, CD133 (also called prominin-1/AC133) has also been used as a marker to enrich normal human HSCs as well as stem cells in the human central nervous system, suggesting that it may be a marker of both normal and malignant stem cells.
Recently, there have been reports that CD133 may not be a universal marker of CSCs in brain cancer.22,23,24 In addition, a study of neurosphere lines derived from PTEN-deficient human glioblastomas found that both CD133+ and CD133− cells could generate serially transplantable tumors in xenograft recipients and provided evidence that these tumors comprise a hierarchy of self-renewing CSC populations with variable tumorigenic capacity.25 Unfortunately, these investigators were not able to study sorted cell populations directly isolated from patient tumors. Cell surface phenotype can change significantly during culture, without corresponding changes in stem cell function.26 In addition, there is evidence that CD133 expression can vary as a function of cell cycle.27 Thus, the results of these studies should be interpreted with caution. Nevertheless, it would not be surprising to find that the CSC phenotype is heterogeneous, even among tumors of the same histologic subtype, given the variety of genetic and epigenetic
perturbations that can ultimately lead to tumor formation. These observations underscore the importance of characterizing candidate CSC populations through functional assays of their tumorforming ability rather than relying solely on phenotypic identification.
perturbations that can ultimately lead to tumor formation. These observations underscore the importance of characterizing candidate CSC populations through functional assays of their tumorforming ability rather than relying solely on phenotypic identification.
Cancer Stem Cells in Other Solid Tumors
Two groups initially reported isolation of CD133+ tumor-initiating cells from human colon cancers.28,29 Single-cell suspensions of primary or metastatic tumor samples were injected either under the renal capsule of NOD/SCID mice28 or subcutaneously into SCID mice.29 In both studies, only CD133+ and not CD133− cells, which composed the bulk of the cancers, were able to initiate tumor formation in vivo. Tumors could be serially propagated by reisolating CD133+ cells from xenografts and transplanting them into secondary mice. The ability to perform quantitative analysis is an essential feature of any in vivo assay. In one study, the frequency of colon CSCs in the bulk tumor was determined by limiting dilution analysis to be 1 in 60,000 colon cancer cells.28 The frequency of CSCs in the CD133+ cell fraction was 1 in 262, representing a greater than 200-fold enrichment over unfractionated cells. Clearly, however, the majority of CD133+ colon cancer cells are not CSCs. As has been shown in the CD34+ cell fraction of AML,30 there may be a hierarchy of CSCs and progenitors in the CD133+ subpopulation of colon cancer cells. Another study identified colon CSCs in the EpCAMhighCD44+CD166+ cell fraction.31 CD44+ cells generally constituted a minority subset of the CD133+ population, thus CD44 is a marker that could potentially be used to purify the CSC-containing fraction further.
Using methodology similar to that described above, CD44+ CSCs have recently been characterized in squamous cell carcinomas of the head and neck (HNSCC),32 adding to the rapidly growing list of human cancers in which a distinct CSC population has been identified (Fig. 11.2, Table 11.1). Significantly, in moderately to well-differentiated HNSCC in which some tissue architecture is preserved, CD44+ cells were localized to the basal layer and costained with Cytokeratin 5/14, a marker of normal squamous epithelial stem and progenitor cells, but not with the differentiation marker involucrin. Furthermore, CD44+ cells expressed much higher levels of BMI1 than CD44− cells. BMI1 plays an important role in the self-renewal of hematopoietic and neuronal stem cells and has been implicated in tumorigenesis.33,34,35 These findings demonstrate that biological pathways likely differ between tumorigenic and nontumorigenic populations and underscore the importance of identifying and characterizing the CSCs within tumors, both for gene expression and proteomic analyses as well as therapeutic targeting.
Expression of CD44 on CSCs from both breast cancer and HNSCC suggests that this adhesion molecule may also be a marker of CSCs in other tumors of epithelial origin. Recently, Li et al.36 showed in pancreatic adenocarcinoma that cell fractions expressing CD44, CD24, epithelial specific antigen (ESA), or a combination of these markers were enriched for tumorigenic activity, as assessed by the frequency of tumor formation following subcutaneous or intrapancreatic injection into NOD/SCID mice. The most highly enriched fraction comprised cells that express all three of these markers, with tumor formation in half of mice receiving as few as 100 CD44+CD24+ESA+ cells. However, injection of CD44−, CD24−, or ESA− cells also gave rise to tumors, albeit with lower frequency. Thus, none of the markers used in this study enabled clear separation of cells with tumor-initiating activity from the bulk nontumorigenic population. In contrast to these results, Hermann et al.37 identified CSCs in pancreatic tumors by CD133 expression; as few as 500 freshly isolated CD133+ cells were able to generate tumors after orthotopic injection into immune-deficient mice, whereas as many as 1 × 106 CD133− cells could not.
The list of tumors in which CSC populations have been identified continues to grow (Table 11.1). There is frequent overlap in cell surface phenotype among CSCs from different tumor types, as evidenced in particular by expression of CD44 and CD133 on CSCs from epithelial tumors. Whether these are simply surrogate markers or play a functional role in CSC biology has yet to be determined.
Controversies and Future Directions
There have been a number of criticisms of the CSC model. As discussed above, characterization of CSC populations requires xenotransplantation into immune-deficient recipients. Some investigators have argued that inefficiencies of the xenotransplant system lead to underestimation of the frequency of cells with tumor-initiating ability.38 In fact, when improvements have been made to these assay systems, for example through the use of more immune-deficient recipients such as NOD/SCID Il2rg−/− mice, the frequency of tumor initiating cells is sometimes dramatically increased, as seen recently for melanoma (greater than 4 log difference).39,40 There are a number of issues to be considered here. First, such large increases in CSC frequency with the use of more immune-deficient recipients are not universal—for example, they have not been observed in AML41,42,42a—possibly reflecting variable sensitivity of tumor-initiating cells from different tumor types to residual host
immune surveillance. Second, while human cell engraftment in xenotransplant assays is undoubtedly limited by residual elements of the recipient immune system, absence of cross-reactivity of cytokines, and other components of the host microenvironment, the low frequency of tumor-initiating cells seen in some cancers cannot be wholly explained by xenotransplantation barriers. For example, LSC frequency in two murine models of leukemia involving MOZ-TIF expression or Pten deletion was low (1 in 104 to 1 in 6 × 105) despite syngeneic bone marrow transplantation.12,43 Furthermore, in contrast to the low frequency of LSCs generally seen when human AML cells are xenotransplanted, much higher LSC frequencies (on the order of 1%) have been observed in a genetically induced model of human B-cell acute lymphoblastic leukemia (ALL) that involves transplantation of human cord blood stem or progenitor cells expressing the MLL-ENL oncogene into immune-deficient mice.42,44 This frequency was comparable to that reported for a murine model of MLL-AF9-induced AML (1 in 150).45 These findings indicate that CSC frequencies can vary widely in different cancers, regardless of whether they are quantified using xeno- or syngeneic transplant assays, and likely relate at least in part to the specific underlying oncogenic pathways operating within the different tumors. Third, and perhaps most important, the CSC hypothesis does not address the absolute frequency of these cells: it is not required that CSCs be rare. The CSC model, at its most fundamental level, simply proposes that the basis of functional heterogeneity within tumors is the existence of cell populations that can be distinguished from other tumor cells by their ability to initiate malignant growth in vivo.
immune surveillance. Second, while human cell engraftment in xenotransplant assays is undoubtedly limited by residual elements of the recipient immune system, absence of cross-reactivity of cytokines, and other components of the host microenvironment, the low frequency of tumor-initiating cells seen in some cancers cannot be wholly explained by xenotransplantation barriers. For example, LSC frequency in two murine models of leukemia involving MOZ-TIF expression or Pten deletion was low (1 in 104 to 1 in 6 × 105) despite syngeneic bone marrow transplantation.12,43 Furthermore, in contrast to the low frequency of LSCs generally seen when human AML cells are xenotransplanted, much higher LSC frequencies (on the order of 1%) have been observed in a genetically induced model of human B-cell acute lymphoblastic leukemia (ALL) that involves transplantation of human cord blood stem or progenitor cells expressing the MLL-ENL oncogene into immune-deficient mice.42,44 This frequency was comparable to that reported for a murine model of MLL-AF9-induced AML (1 in 150).45 These findings indicate that CSC frequencies can vary widely in different cancers, regardless of whether they are quantified using xeno- or syngeneic transplant assays, and likely relate at least in part to the specific underlying oncogenic pathways operating within the different tumors. Third, and perhaps most important, the CSC hypothesis does not address the absolute frequency of these cells: it is not required that CSCs be rare. The CSC model, at its most fundamental level, simply proposes that the basis of functional heterogeneity within tumors is the existence of cell populations that can be distinguished from other tumor cells by their ability to initiate malignant growth in vivo.
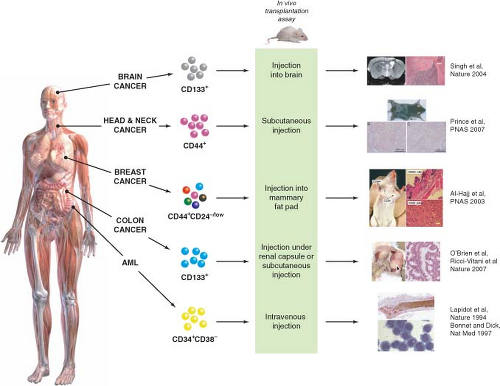 FIGURE 11.2 Identification of cancer stem cells (CSCs) in acute myeloid leukemia (AML) and solid tumors. Subfractions of tumor cells isolated from the bulk tumor population are assayed for their ability to initiate tumor growth in vivo using immune-deficient mice as recipients. CSCs are the only cells capable of initiating tumor growth, giving rise to more CSCs through self-renewal, and also to nontumorigenic differentiated progeny, thus recapitulating the functional heterogeneity of the original tumor. CSCs were first identified in AML and have now also been identified in several types of solid tumors (Table 11.1). (Brain cancer images adapted by permission from Macmillan Publishers Ltd: Nature (21), copyright 2004. Head and neck cancer and breast cancer images copyright 2007 (ref. 32) and 2003 (ref. 15), respectively, National Academy of Sciences, USA. Colon cancer images are from ref. 28.) |
It is possible that some human cancers may not follow the CSC model. For example, the high frequency (one in four) and lack of a clear isolatable phenotype of tumor-initiating cells in melanoma suggest the absence of a hierarchical organization.40 Regardless of whether a tumor contains a subpopulation of CSCs responsible for maintaining tumor growth or common tumorigenic cells with little evidence of a CSC hierarchy, all cancer cells with the ability to initiate disease must be identified and targeted therapeutically in order to achieve cure. Since the first published reports of CSCs in breast and brain cancers, there have been numerous studies of CSCs in other solid tumors, with varying degrees of robustness. When weighing evidence presented for or against the CSC hypothesis, one must consider not only the rigor of the xenotransplant assay but also experimental details such as whether cancer cells have been extensively cultured in vitro or passaged in vivo, both of which can lead to changes in function and phenotype, and whether freshly isolated cells from patients’ tumors have been used versus cell lines that may not be representative of clinical disease.
TABLE 11.1 PROSPECTIVE ISOLATION OF CANCER STEM CELLS FROM HUMAN LEUKEMIAS AND SOLID TUMORS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|