The past 25 years have seen an increase in our understanding of immunology and further expansion in the clinical use of immunotherapeutic modalities. How immunotherapy will be integrated with chemotherapy, radiation, and surgery remains to be established. Although there have been successes in the field of immunotherapy, they have been inconsistent, and it is hoped that increased understanding of the basic principles of immunology will improve the consistency of beneficial effects. In this article, we briefly provide a general overview of our current understanding of the immune system, with a focus on concepts in tumor immunology, followed by a discussion of how these concepts are being used in the clinic.
The initial use of immunotherapy for cancer occurred in the early 1900s when Coley used bacterial products to treat patients who had Ewing sarcoma based on the observation that postoperative infections seemed to diminish the likelihood of tumor recurrence. A number of patients were treated with these bacterial products, resulting in regression in a few. James Ewing was simultaneously testing radiation as a means to treat these sarcomas, and controversy as to which approach was superior ensued. The consistency in response seen with radiation led to this treatment being more widely accepted, and the field of immunotherapy would need to wait approximately 50 years until it was explored further. The past 25 years have seen an increase in our understanding of immunology and further expansion in the clinical use of immunotherapeutic modalities. How immunotherapy will be integrated with chemotherapy, radiation, and surgery remains to be established. Although there have been successes in the field of immunotherapy, they have been inconsistent, and it is hoped that increased understanding of the basic principles of immunology will improve the consistency of beneficial effects. In this article, we briefly provide a general overview of our current understanding of the immune system, with a focus on concepts in tumor immunology, followed by a discussion of how these concepts are being used in the clinic. Although this overview illustrates the highly integrated nature of the immune system, we divide the clinical section into specific arms of the immune response. It is likely that, as with the natural immune response, immunotherapy is most effective when the components of the immune armamentarium are used in combination.
Principles of the immune response
The immune system evolved to protect the host from invading pathogens. These processes can effectively clear aberrant self-antigens, including malignant cells. A complete description of the immune response is beyond the scope of this article, but we highlight areas relevant to cancer immunotherapy. In general, the immune system can be divided into the innate response, which allows rapid, nonspecific protection, and the adaptive response, which develops more slowly but provides specific recognition of antigens via expression of carefully rearranged receptors. Fig. 1 illustrates the various components of the immune response. Although the innate system is an essential part of successful immune clearance, this article focuses on adaptive immunity.
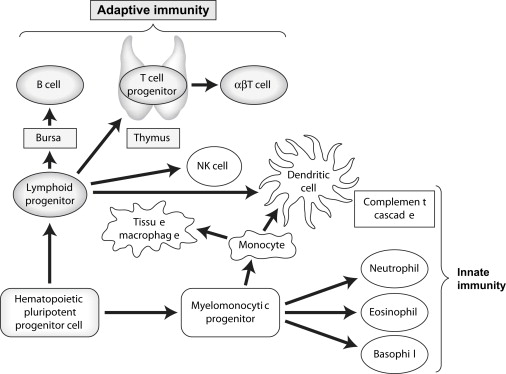
The adaptive immune system contains millions of potential specificities requiring amplification upon initial antigen encounter, which results in delayed onset but allows for a memory effect such that subsequent exposures to antigen result in a more rapid clearance. One potential danger of this type of system is the recognition of self-antigens resulting in autoimmunity. To prevent this, a number of strategies have evolved, including central deletion of specificities directed against self-antigens (in the thymus for T cells); the development of a complex network of regulatory cell types that maintain tolerance to self-antigens in the periphery (outside the thymus); and the requirement for professional antigen-presenting cells, such as dendritic cells (DCs), to effectively initiate the adaptive immune response. DCs are capable of sensing, filtering, and interpreting signals for the adaptive immune cells, providing an important link between the innate and adaptive immune response.
T Cells and B Cells
Developing T cells and B cells rearrange germline DNA to generate receptors that are maintained throughout subsequent progeny and are capable of recognizing specific antigens. For B cells, gene rearrangement initially occurs in the bone marrow, with further rearrangements possible in the germinal center of the lymph node during maturation of the immune response after antigen encounter. For T cells, this process occurs in the thymus, where specificities are selected on the basis of recognition of self–human leukocyte antigens (HLAs) followed by deletion of T cells with high affinity for self-antigens. Although this process is complex and results in loss of greater than 95% of all rearranged T cells in the thymus, it allows for sufficient repertoire to protect throughout the lifespan of the host without causing autoimmunity in most individuals. Naive T cells and B cells circulate in a resting state until they encounter an antigen that binds to the specific receptor expressed on the surface of a responding cell. For T cells, this antigen is presented in the context of self HLA molecules on specialized antigen-presenting cells (APCs), such as DCs. CD4+ T cells recognize antigen in the context of HLA class II molecules, and for CD8+ T cells this recognition occurs on HLA class I molecules. Effective stimulation of the immune response also requires a second costimulatory signal provided by the APCs. The nature of a costimulatory signal (positive, negative, and how strong) is modulated by factors sensed by the APCs, such as bacterial products or inflammatory substances produced during the innate immune response (also referred to as “danger signals”. ) An important group of receptors known as toll-like receptors (TLRs) recognizes these products and modulates the capacity for the APCs to stimulate a T-cell response, thus demonstrating one important link between innate and adaptive immunity. Once activated, T and B lymphocytes undergo rapid clonal expansion, providing large numbers of effectors. T cells mediate an immune response via direct cytotoxicity of the target cell (perforin, granzyme, fas/fasL) or by secretion of effector cytokines, such as interferons, whereas B cells differentiate into antibody-secreting plasma cells. T and B lymphocytes also “talk” to other immune cells (innate and adaptive) by secretion of cytokines or through expression of surface molecules, resulting in further refinement in the immune response. Cytokines such as interleukin (IL)-2, IL-7, or IL-15 have been administered in clinical and preclinical studies to enhance antitumor immune responses. In addition, agents that interfere with regulatory signals generated by surface receptors on responding immune cells, such as CTLA-4 (a negative regulator of T-cell immunity), have been used in patients who have cancer and who have demonstrated clinical activity. Fig. 2 demonstrates a schematic of the immune response.
An important subset of T cells generated in the thymus acquires a regulatory phenotype before export into the periphery, providing further protection against autoimmunity. These naturally occurring regulatory T cells (Tregs) can be identified by expression of CD4 and CD25 (a component of the IL-2 receptor), low or absent expression of the IL-7 receptor, and the FoxP3 transcription factor. There is a subset of Tregs that can be induced during an immune response that may express the CD4 or CD8 co-receptor. Our growing understanding of Tregs has led to the recognition that these cells may be detrimental to an effective antitumor immune response, particularly when the antigens targeted are self-antigens for which these regulatory networks are well developed. Current immunotherapeutic protocols are exploring depletion or the modulation of Tregs as a means to enhance adaptive immune responses.
Natural Killer Cells
Natural killer (NK) cells are bone-marrow–derived lymphocytes that do not bear clonally rearranged antigen-specific receptors. In humans, NK cells are phenotypically defined as CD3 − /CD56 + lymphocytes, which can be divided into CD56 dim (CD16 + ) cytotoxic and CD56 bright immunoregulatory NK-cell subsets. NK cells have the potential to recognize and eliminate a wide range of tumors and virally infected cells. Besides acting as cytotoxic effector cells in immune responses, evidence is accumulating that NK cells play a crucial role in the translation of signals from innate toward adaptive immunity via bidirectional interaction with DCs. Recent evidence indicates that type I interferon (IFN)-experienced DCs prime NK cells in an IL-15–dependent manner. Primed NK cells can promote maturation of DCs in an IFN-γ–dependent manner, thereby facilitating further induction of T H1 (cellular) T-cell responses. Therefore, NK cells may exert a dual function in antitumor responses by acting as direct effector cells and as initiators of T-cell–mediated antitumor responses.
The functional status of NK cells is regulated by the balance of inhibitory and activating NK-cell ligands on the target cells. Inhibitory signals are provided by classical and nonclassical HLA class I molecules, which bind to killer immunoglobulin- like receptors (KIRs) or NKG2A/CD94 on NK cells. Within the human population, there is a wide variation in the repertoire of KIR genes. Together with the clonal distribution within the NK repertoire, this results in a wide diversity of expression profiles between and within individuals.
The inhibitory signals can be decreased in patients who have solid tumors caused by a down-regulation of HLA class I on the tumor cells (see “Immune escape”). Lack of HLA-dependent inhibitory signals may permit recognition and elimination of tumor cells by NK cells according to mechanism of “missing self” recognition. In addition, interaction between NK-cell–activating receptors and their specific ligands on target cells seems necessary for adequate NK cell stimulation. NK cells can be triggered by stress-induced ligands that are expressed by the tumor itself, such as the MIC (MICA/B) and ULBP (ULBP1-4) family of proteins. The expression of these molecules on tumors can be induced by DNA damage. The activating receptor NKG2D on the NK cell can bind to these ligands and lead to an activating signal. Other activating NK cell receptors include the DNAX accessory molecule-1, which is activated by the CD112 (Nectin-2) and CD155 (PVR) molecules. A third group of activating receptors is represented by the natural cytotoxicity receptors NKp30, NKp44, and NKp46. Their physiologic ligands remain to be identified.
Many preclinical studies have provided evidence that a broad spectrum of human and murine tumor cell lines are susceptible to NK-cell cytotoxicity, albeit with different efficacy. The variation in NK-cell susceptibility seems related to differences in expression levels of the aforementioned activating and inhibitory ligands on tumor cells. In addition to tumor cell lines, NK cells have been shown to have the potential to eliminate or prevent outgrowth of murine tumors in in vivo models. In humans, the clinical significance of NK cells in the control of solid tumors is unresolved and is the subject of many studies.
NK-cell activation and cytolytic potential can be increased upon stimulation with various cytokines (eg, IL-2, IL-12, and IL-15) and type I IFN. In various clinical trials, in vivo administration of some of these agents has been used to induce NK-cell–mediated antitumor responses with limited clinical efficacy. Further insight in the role of these agents in NK cell regulation will provide tools to manipulate NK-cell function and will be of benefit in future adoptive transfer studies.
Another dimension of NK tumor target recognition may be seen in an allogeneic setting where a KIR–HLA mismatch between donor and patient may result in the absence of tumor cells of ligands for inhibitory KIR on donor NK cells. In this setting of “missing self-recognition,” the available alloreactive donor NK-cell repertoire may be exploited to eliminate recipient tumor cells. Combined with the preferential expression of activating NK-cell ligands on malignant cells, this might result in a selective elimination of these targets without the risk of inducing concurrent graft-versus-host disease (GvHD). The first evidence for the clinical implication of this model has been reported by Ruggeri and colleagues in patients who have acute myeloid leukemia undergoing haploidentical stem cell transplantation. In their experience, KIR-HLA mismatch was associated with a better clinical outcome, lower relapse rate, and reduced frequency of GvHD. In subsequent studies, conflicting results have been reported on the advantage of KIR-HLA that may be related to the level of T-cell depletion and the repertoire of alloreactive donor NK cells. Further insight into reconstitution, regulation, and functional properties of the (alloreactive) NK-cell repertoire after allogeneic stem cell transplantation (SCT) is needed to optimize exploitation of its potential. Altogether, the mode of action of NK cells indicates that they have the potential to exert potent antitumor immunity and may act complementary to and in synergy with T cells.
Immune Escape
As first proposed by Burnett, tumors are not passive targets for cellular immune responses and are capable of escaping from and disabling the host immune system. Changes in tumor phenotype not only permit tumor escape from normal immunologic surveillance but may also negatively influence the susceptibility and response to antitumor immunotherapy. In the setting of clinically established tumors, initial immune surveillance has failed to eliminate tumor cells. Immune pressure may have resulted in the generation of immune escape variants via the mechanism of immuno-editing. General mechanisms of immune escape include interference with specific recognition of tumors by the immune system, reduced susceptibility of tumors to the apoptosis-inducing capacity of cytotoxic effector cells, and immunosuppressive potential of the tumor. These mechanisms have been described in detail elsewhere and are therefore only be briefly discussed here.
Immune Cell Recognition of Tumors
Appropriate expression of HLA/peptide complexes on tumor cells plays a crucial role in the effector phase of cellular immunotherapy because they present antigenic epitopes to CTLs. A commonly known mechanism of immune escape involves impaired antigen processing or presentation. Total or selective loss of HLA I expression has been reported in a large variety of human tumors, including sarcomas, carcinomas, and lymphomas. The clinical relevance has been demonstrated by the observation that a lack of HLA expression often relates to metastatic disease and an unfavorable outcome. The basis of aberrant HLA expression has been reported to be genetic, regulatory, or epigenetic in origin. Lack of HLA expression in tumors may be caused by loss of heterozygosity, mutations, or deletions in individual HLA genes and in genes encoding β2 microglobulin and components of the antigen-processing machinery, including peptide transporters TAP1 and TAP2, tapasin, and the proteosomal subunits LMP-2 and LMP-7. The aberrant HLA expression pattern in tumors at diagnosis, which may be heterogeneous within individual tumors, indicates that a process of selection pressure has occurred. Evidence that immune surveillance and immune pressure may result in selective outgrowth of tumor variants has come from observations in clinical trials in which progressive disease after cellular immunotherapy revealed total or allelic loss of HLA expression.
Recently, a unique category of CTLs has been identified in mice bearing TAP-deficient tumors. These tumors were found to express peptides derived from self-antigens in the context of classical and nonclassical MHC class I molecules, named T-cell epitopes, associated with impaired peptide processing, which may serve as alternative tumor-specific CTL targets. Together with conventional tumor-specific T cells, T cell epitopes associated with impaired peptide processing (TEIPP)-specific CTLs might be considered in future studies to prevent outgrowth of or treat TAP-deficient tumors.
In addition to genetic causes, aberrant HLA expression may be the result of transcriptional regulation. During normal immune responses, HLA class I and II expression is strictly orchestrated and mediated by several immune regulators, including IFN-γ and TNF-α, that have the potential to increase HLA expression in a variety of HLA-deficient tumor cell lines. Another possible mechanism responsible for transcriptional regulation of HLA expression is represented by epigenetic modifications, including histone acetylation and DNA methylation. Treatment with histone deacetylase inhibitors and methyltransferase inhibitors has been demonstrated to restore HLA expression in a variety of tumors and hematologic malignancies. Whether this category of agents, which has been used as anticancer therapy in clinical trials, may be beneficial in future clinical immunotherapy studies remains to be resolved.
Tumors may specifically down-regulate expression of CTL tumor target antigens, which result in antigenic loss variants. The clinical significance of this mechanism has been clearly observed in immunotherapy trials with patients who have melanoma in whom metastases occurring after treatment were found to selectively lack expression of CTL target antigens. This illustrates the biological mechanism that selective pressure has the potential to generate immune escape variants. In addition, it shows that antigen-specific therapies, although elegant, may be vulnerable to the process of immune editing, especially in cases where tumor target antigens are used that are not essential for tumor biology and survival. Another potential hurdle for antigen-specific therapy is the often heterogeneous expression of target antigens, such as cancer-testis antigens, within individual tumors. Epigenetic regulation has been found to play a significant role in the expression of these genes, and treatment with the aforementioned hypomethylating agents has the potential to restore or increase expression, which may be beneficial in future immunotherapy studies.
Although partial or complete loss of HLA/peptide complex expression impairs T-cell–mediated recognition, it may increase susceptibility to NK cells, which may be inhibited by self-HLA (see section on NK cells). Aberrant expression on the tumor of the ligands for NK-cell–activating receptors or expression of HLA-surrogate molecules can protect tumor cells from NK-cell–mediated recognition and killing. Several mechanisms leading to this so-called “NK-cell tolerance” have been described. First, sustained expression of its ligand induced down-regulation of NKG2D in a murine tumor model, resulting in increased tumorigenesis. This process seemed to be reversible by stimulating innate immunity through TLRs. Similarly, expression of natural cytotoxicity receptors was found to be reduced in patients who have acute myelogenous leukemia (AML) compared with healthy control subjects, which was in part reversible by cytokine stimulation. Second, NKG2D expression and consequent NK-cell (but also T-cell) function may be blocked and thus impaired by tumor-derived soluble ligands, as reported for soluble MICA in various human tumors. Third, expression of the nonclassical HLA-E and G molecules on tumors has the potential to inhibit NK-cell function via interaction with their respective receptors. A fourth potential mechanism is the aberrant expression of NK co-receptors and (soluble) adhesion molecules, which may prevent a functional interaction.
Tumor Susceptibility to Immune Cell-mediated Killing
Interference with the induction of apoptosis is a frequently observed phenomenon in tumors and may be caused by a variety of mechanisms. This article focuses on the mechanisms directly related to the mode of action used by cytotoxic effector lymphocytes and particularly those reported in human tumors. Tumor cells may interfere with the cytolytic pathways used by NK and T cells by overexpression of antiapoptotic genes or by down-regulation of proapoptotic molecules.
Overexpression of serine protease-inhibitor 9 has been shown to irreversibly inactivate GrB, resulting in defective apoptosis in tumor cell lines via the granule exocytosis pathway. The clinical significance has been suggested by the correlation between protease inhibitor–9 overexpression and clinical outcome in lymphomas and the outcome after tumor vaccination in patients who have melanoma. Escape from death receptor (DR)-mediated apoptosis by overexpression of cellular FLICE inhibitory protein, which is a catalytic inactive homolog of procaspase 8, has been demonstrated in various types of tumors in vitro and in vivo. Negatively affecting the same pathway, lack of caspase 8 expression has been reported to interfere with DR-mediated apoptosis in several human tumors and to favor formation of metastases.
Other possible mechanisms to escape from DR pathway–mediated apoptosis include the expression of soluble (eg, soluble CD95 ); and decoy receptors (eg, DcR 3 ); and the presence of mutations in proteins involved in the DR cascade that may interfere with DR-mediated apoptosis. Although the presence of these mechanisms was found to be correlated with clinical outcome of various tumors, their direct involvement in escape from immune pressure in vivo is remains to be resolved.
Tumor-induced Immune Suppression
It has been extensively reported that cancer patients are often characterized by a general state of decreased immune competence by mechanisms that are only partially understood. These mechanisms have been described in detail in several recent reviews. Therefore, only a selection of mechanisms actively induced by the tumor is mentioned here. First, tumors may actively secrete immune suppressive cytokines (eg, IL-10, TGF-β and Indolamine 2,3-Dioxygenase [IDO]) that are able to interfere in various ways with innate and adaptive immune responses. Second, an increased frequency of circulating Tregs and their migration to the tumor environment has been reported in cancer patients. The influx of Tregs in tissue or the surrounding stroma varied among different tumor types and between individuals. An association between Tregs numbers and clinical behavior (progressive disease) was first described in ovarian cancer patients. The immunosuppressive effect induced by Tregs involves several potential mechanisms mediated via soluble factors and direct cell–cell contact as described in the previous sections on T-cell–mediated responses. Third, tumors have the potential to attract myeloid suppressor cells that may exert multiple inhibitory functions, including suppression of tumor infiltrated lymphocytes. Fourth, down-regulation of T-cell receptor signaling molecules that may be reversible upon removal of the tumor has been reported in cancer patients.
Lymphocyte Migration
For cellular immunotherapy to be effective, migration of effector T or NK cells to the tumor site, extravasation, and invasion into the tumor are pivotal. The presence of and variability in the amount of tumor-infiltrating lymphocytes (TILs) have been reported in many human tumors. In some studies, the presence of these TILs was reported to be correlated with the pattern of HLA expression. Recent studies in colorectal, ovarian, and cervical tumors have shown that the phenotypical profile of the TIL at diagnosis was a strong predictor of clinical outcome. Particularly, the CD8/Treg ratio was found to be positively correlated with a favorable prognosis. These findings illustrate that insight into the molecular mechanisms involved in tumor site–directed migration are important to the understanding of the quantitative and qualitative differences in the naturally occurring TIL responses. This provides knowledge required to develop strategies to manipulate the natural immune response and to support (adoptive) immunotherapy interventions.
Site-directed migration of T/NK cells is a nonrandom process induced by inflammatory and other pathogenic stimuli. In lymphoid and in inflamed nonlymphoid tissues, lymphocyte adherence and tissue influx are facilitated by specialized high-endothelial venules (HEVs). HEVs express various adhesion molecules (eg, VCAM-1 and ICAM-1) and chemokines (eg, CCL21, CXCL9, and CXCL25), which create a highly regulated interface between lymphocytes and endothelial cells. In contrast, intratumoral vessels are characterized by squamous endothelial cells with low expression of these molecules. Generally, lymphocyte extravasation at the tumor site is limited. Significant differences have been reported between intratumoral- (low) and peritumoral- (dense) vessel lymphocyte extravasation. The molecular basis for these differences remains to be resolved, but a regulatory role has been suggested for pro- and antiangiogenic factors (eg, vascular endothelial growth factor), anti-inflammatory cytokines (eg, TGF-β), and the tumor cells themselves. Transformation of squamous “nonattractive” tumor vessel endothelium into endothelial cells with a HEV-like appearance could have a beneficial effect on lymphocyte migration at the tumor site. Potential approaches include stimulation with inflammatory mediators (eg, TLR ligands), ionizing irradiation, and transgenic expression of recruiting cytokines.
Chemokines are a superfamily of small molecules that regulate this selective process of migration. Directional migration of T/NK cells expressing the appropriate chemokine receptor(s) occurs along a chemical gradient of ligand(s). In cancer, chemokines produced by the tumor may play a role in the pattern of leukocyte infiltration. Several recent studies on human tumors have provided evidence for the significance of tumor-secreted chemokines on TIL responses and clinical outcome. Immune stimulatory and inhibitory effects have been proposed depending on the extent and by which cells the chemokines are produced in the tumor environment. Identification of the relevant chemokines involved in the attraction of cytotoxic effector T or NK cells and Tregs combined with insight in the patterns of expression and regulation of these chemokines in human tumors may provide tools to manipulate the attractive capacity of the tumor and thereby improve the process of tumor-site–directed migration during T/NK-cell–mediated immunotherapy studies.
Clinical experience with immune-based therapies for cancer
T-cell Therapy
A number of strategies have been developed to use T cells as immunotherapeutic tools against tumors. Adoptive therapies involve the infusion of large numbers of T cells into patients (autologous or allogeneic). Vaccine therapy attempts to expand T cells, recognizing tumor associated antigens in vivo. Finally, cytokines have been used alone or combined with other strategies to expand or enhance the function of antitumor T cells.
Autologous
The use of autologous T cells to target malignancy can involve the infusion of manipulated or unmanipulated T cells or the administration of vaccines to expand tumor-reactive T cells in vivo. The existence of tumor-specific or tumor-associated antigens that can be targeted using these approaches has been clearly demonstrated. The majority of these antigens are self-antigens expressed during a restricted period of development or in restricted tissues. Therefore, the process of inducing effective antitumor T-cell immunity requires that the self-tolerance mechanisms previously described be overcome. This has presented one of the major obstacles to effective T-cell–based therapies.
For adoptive T-cell therapies, the source can be peripheral blood T cells or tumor-infiltrating T cells, which can then be harvested and reinfused into patients. T cells reactive against tumor antigens are present but infrequent in the blood of cancer patients. Although these T cells may be present at higher frequency in tumor infiltrates, the total number of cells that can be harvested is insufficient. Thus, effective adoptive immunotherapy requires manipulation and expansion in vitro to increase the frequency of T-cell–recognizing tumor antigens in the infused product. This approach has been used in the clinic and has resulted in regression in patients who have melanoma. A number of other clinical trials using adoptive immunotherapy have been undertaken, some demonstrating clinical benefit. The experience of Dudley and colleagues suggested that regression required that extremely high doses of T cells be infused such that a high percentage of circulating T cells recognized the tumor (∼20–30%). These important findings indicate proof of the principle that, at least for melanoma, adoptive T-cell therapies represent a promising immunotherapeutic modality. An alternative method to overcome the low frequency of tumor reactive T cells in autologous products that is being explored is gene transfer of T-cell receptors known to recognize tumor antigens into cells with cytotoxic potential, such as T cells or NK cells. Although there has been much progress in the area of autologous adoptive immunotherapy for cancer in adults, there are limited data in pediatric patients.
Another approach that can be used alone or in combination with adoptive T-cell therapy is the use of vaccines to expand tumor-reactive T cells. The types of vaccines used include whole tumor cells, peptides derived from known tumor antigens, replication-deficient viruses expressing tumor antigens, and DCs loaded with tumor antigens. A large and growing number of clinical trials have been undertaken using each of these vaccine strategies resulting in vaccine responses and some evidence of clinical response, but the potency and consistency of these responses has been poor. Most of these trials have been undertaken in the setting of bulky tumors where immune-based therapies may be less effective due to the tumor suppressive mechanisms discussed previously. Furthermore, the magnitude of the immune response generated by vaccines alone suggests that combining vaccines with adoptive therapies or T-cell–active cytokines may be necessary. Another strategy is the use of adjuvants to amplify weak vaccine-induced T-cell responses. There is the most clinical experience with Freund’s adjuvant, but newer agents are being explored that specifically target innate immune cells via TLRs. It is likely that effective vaccination protocols will need to incorporate multiple strategies to generate sufficient immune responses to induce clinically meaningful responses. As with adoptive immunotherapy, experience in pediatrics is limited, but there has been one promising clinical response in pediatric sarcoma.
Allogeneic
Perhaps the most potent form of immune-based therapy is the graft-versus-tumor reaction that occurs after allogeneic transplantation. T cells and NK cells contribute to this response. The T-cell contribution is evident from the increased risk of relapse that occurs after transplantation of stem cell products that are depleted of T lymphocytes and when patients are treated with T-cell immunosuppressants. For chronic myeloid leukemia (CML) that recurs after transplant, remissions can be induced in up 50% or more of patients by stopping immunosupression or infusing donor lymphocytes (DLI). For AML, responses to donor lymphocyte infusions occur, but the frequency of responses is substantially lower than for CML (∼20–30%). For pediatric acute lymphocytic leukemia, 5-year disease-free survival after allogeneic transplantation ranges from approximately 40% to 80%, depending on risk status and type of donor used. If relapse occurs, the poor response to immune manipulation suggests that the graft-versus-leukemia effect is far less potent than for AML or CML, although the reasons for this are not clear. Acute lymphocytic leukemia blasts are inferior at initiating immune responses but are susceptible to autologous T-cell–mediated immune responses. Thus, for the most common form of pediatric leukemia, strategies to enhance the graft-versus-leukemia response are needed if outcomes are to be improved.
The effectiveness of the graft-versus-malignancy response is, to a large extent, due to the ability to target minor histocompatibility antigens that are disparate between the donor and recipient because these antigens do not require the immune response to overcome tolerance to self-antigens. Another advantage of allogeneic transplant is the availability of donor-immune cells that have not been depleted or exposed to cytotoxic therapy and are not contaminated by tumor cells. Finally, it is possible that tumor restricted immune responses induced in the allogeneic transplant environment may be more potent than similar responses induced using the autologous environment. Thus, there are a number of potential benefits in considering allogeneic transplantation as a platform for immune-based therapies.
The main hurdle to overcome when using allogeneic transplantation as immunotherapy is the induction of GvHD. Most of the available approaches to enhance graft-versus-tumor reactions are nonspecific, such that the antitumor reaction is closely linked to GvHD. The use of strategies such as vaccines or adoptive therapy with antitumor T-cell–enriched donor lymphocytes may be potential mechanisms to overcome this hurdle. An alternative would be to develop strategies to selectively modulate alloresponses against GvHD target organs. For example, selective depletion of alloreactive T cells in vitro before infusion of stem cells has been explored in preclincical and clinical settings. The ability to manipulate the post-transplant environment to enhance the graft-versus-malignancy reaction would result in less reliance on pretransplant conditioning for cure, potentially allowing the use of reduced-intensity conditioning regimens, which have been used successfully for pediatric nonmalignant diseases and adult malignancies. Given the long-term morbidity associated with myeloablative transplantation for pediatric patients, this would be a desirable scenario.
Natural Killer Cell Therapy
Autologous
The first studies in this field were performed in the 1980s by the Rosenberg team. Autologous IL-2/lymphokine-activated killer cells, combined with high-dose IL-2 in vivo, were used to treat cancer patients who had refractory disease, including melanoma and renal cell carcinoma.
In subsequent years, many more patients were included in similar studies receiving lymphokine activated killer cells combined with IL-2 or IL-2 alone, resulting in a response rate of 10% to 20%. The limited efficacy and substantial toxicity of this approach together with the identification of tumor antigens as targets has shifted attention to T-cell–mediated strategies. Progress in our understanding of NK-cell biology and the aforementioned implications of KIR-HLA (mis)matching in allogeneic SCT have resulted in renewed attention for the clinical application of allogeneic NK-cell–mediated immunotherapy strategies.
Allogeneic
The use of haploidentical NK cells in a nontransplant setting was first explored by Miller and colleagues in patients who had refractory hematologic malignancies and solid tumors. For this purpose, NK-cell preparations were obtained from leukapheresis products using immunomagnetic CD3 depletion followed by overnight IL-2 stimulation. From this and subsequent studies, they concluded that a high-dose cyclophosphamide/fludarabine regimen was required to obtain long-term survival and expansion of the infused cells. In addition, NK-cell expansion was found to be correlated with endogenous IL-15 levels, which is in agreement with its role in survival and homeostatic proliferation. Infusion of the NK-cell products did not result in GvHD or other toxicity events. Clinical efficacy was demonstrated by achievement of a CR in a subgroup of patients who had AML. This was only observed in the patients receiving high-dose NK-cell treatment and was correlated with KIR–ligand mismatch in the GvHD direction. This and other pilot studies have provided evidence for the safety and feasibility of allogeneic NK-cell infusions. Further studies are required to investigate the antitumor efficacy in vivo and to unravel the relevant mechanisms involved. In addition to primary NK cells, adoptive transfer studies have been performed using the NK cell line NK-92, expressing activating receptors and lacking inhibitory receptors. The use of these cells was found to be safe, and antitumor responses have been observed.
Several important issues remain unresolved and require further study to optimize the clinical use of allogeneic NK-cell preparations. One of these issues includes the technical approach used to obtain a defined number of NK cells from leukapheresis products and to reduce the amount of contaminating alloreactive T cells. Several procedures, including negative (ie, T and B cells) and positive (ie, CD56) selection steps, which are available under clinical grade conditions, are being investigated by several groups. Second, the absolute number and functional status of NK cells required for efficacy needs to be established. Little is known about dose-response ratios and the in vivo behavior of infused NK cells, but it is likely that this is influenced by multiple factors. Miller and colleagues have demonstrated that a leuko/lymphopenia-inducing preparative regimen seems required to permit engraftment and expansion of adoptively transferred NK cells. Substantial evidence obtained from in vitro and in vivo studies indicates that the functional properties and survival of NK-cell populations, including responses toward tumor cells, can be increased after cytokine stimulation. This seems to justify the preferential use of ex vivo–stimulated NK-cell populations. Endogenous production of cytokines that influence NK-cell activation and survival (eg, IL-15) could play a significant role and is probably dependent on the preparative regimen, timing of NK-cell infusion, and postinfusion therapeutic regimens. By definition, the NK-cell repertoire is phenotypically and functionally heterogeneous, implying that significant interindividual differences will probably be encountered when these cells are used in adoptive transfer studies. In the allogeneic KIR-ligand mismatched setting, the interindividual variability in the amount of alloreactive NK cells is an additional factor that may influence outcome. The challenge is to obtain further insight in the impact of all these parameters on clinical immune and antitumor responses in the scheduled and ongoing clinical trials. Given the reported favorable outcome of KIR-ligand mismatched haploidentical SCT in patients who have refractory AML and the apparent safety profile of allogeneic NK-cell infusions, it seems interesting to investigate exploitation of the NK-cell effect in patients who have NK-cell–permissive solid tumors in a similar haploidentical setting.
B Cells
Antibody therapy
Although this review has emphasized cellular therapies, the use of monoclonal antibodies targeting malignancy is rapidly expanding. Although the majority of these antibodies has been developed for adult cancers, some have demonstrated utility in pediatric malignancy. For example, anti-CD20 has been used in pediatric lymphomas. Although this agent may theoretically target CD20-expressing pediatric B-cell leukemias, responses in this setting have been less promising. One difficulty with monoclonal antibodies alone is that, unless they interfere with receptor signaling on which the tumor is dependant, they require a mechanism such as antibody-dependant cytotoxicity to clear tumor cells. To overcome this, a newer generation of antibodies has been conjugated to radioisotopes or toxins to deliver these directly to the tumor. A number of these antibodies are in clinical trials in pediatric malignancies.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


