10 About 99% of the total body calcium is found in the skeleton bound to phosphate and hydroxyl ions in the form of hydroxyapatite. The normal total serum calcium concentration at all ages ranges from 2.2 to 2.6 mmol/L and consists of physiologically active ionized calcium (50%), with the remainder being either bound principally to albumin or globulins (40%), or circulating complexed to citrate, phosphate, or other constituents in the serum (10%). Calcium in intracellular and extracellular fluid is involved in various metabolic processes, including many enzymatic reactions, hormone secretion, and blood coagulation. Calcium metabolism is regulated primarily by vitamin D, parathyroid hormone (PTH) and calcitonin and serum concentrations are influenced by: Approximately 85% of body phosphate is contained within bone hydroxyapatite. Many cellular reactions require either organic or inorganic phosphate. Normal inorganic serum phosphate concentrations drop from 1.3–2.3 mmol/L in infancy to 0.8–1.5 mmol/L at the end of puberty. About 15% of circulating phosphate is protein bound. Free phosphate is required together with ionized calcium for normal bone mineralization. Vitamin D is either ingested in the diet or synthesized in the skin following ultraviolet irradiation from sunlight. Circulating vitamin D (Figure 10.1) is metabolized in the liver to 25‐hydroxyvitamin D and then in the kidneys to either a metabolically active form (1,25‐dihydroxyvitamin D) or an inactive form (24,25‐dihydroxyvitamin D). 1,25‐dihydroxyvitamin D synthesis is stimulated by hypocalcaemia, PTH and hypophosphataemia. Figure 10.1 Vitamin D metabolism. Circulating vitamin D exerts its target organ effect by binding to an intracellular vitamin D receptor. Vitamin D stimulates the activity of osteoclast‐like cells but suppresses that of osteoblast‐like cells and, in the presence of PTH, mobilizes calcium from bone. 1,25‐dihydroxyvitamin D stimulates intestinal calcium absorption but whether it influences renal handling of calcium and phosphate is less clear. Vitamin D receptors have been located in many other tissues in the body, suggesting a wider role for vitamin D than just regulation of calcium metabolism. The PTH gene has been localized to the short arm of chromosome 11. PTH is synthesized as a pre‐pro‐hormone in the four parathyroid glands. Pre‐pro‐PTH is converted to pro‐PTH as it is transported across the rough endoplasmic reticulum and is stored in secretory granules in the form of the mature 84 amino‐acid peptide PTH. PTH release is stimulated by hypocalcaemia and inhibited by hypercalcaemia acting through a specific calcium‐sensing (G‐protein coupled) receptor on the plasma membrane of the parathyroid cell. This has the capacity to sense small changes in circulating calcium concentrations and to couple this information to intracellular pathways that modify PTH secretion. The primary function of PTH is to prevent hypocalcaemia. Within minutes, changes in PTH secretion affect renal tubular function, increasing calcium absorption and phosphate excretion, and osteoclastic bone resorption. Over a period of one to two days, by stimulating the synthesis of 1,25‐dihydroxyvitamin D, PTH also increases intestinal calcium absorption. PTH produces its target cell effect by binding to a membrane‐bound receptor which stimulates guanine nucleotide‐binding protein (G‐protein) mediated production of cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). This, in turn, stimulates activation of protein kinase A and phosphorylation of intracellular enzymes leading to the physiological action of PTH. Calcitonin is produced in the ‘C’ or parafollicular cells of the thyroid gland. Calcitonin is encoded by a gene also located on the short arm of chromosome 11 and is synthesized in the form of a large precursor molecule. Tissue‐specific processing may lead to an alternative calcitonin gene product, calcitonin gene‐related peptide, which is a potent vasodilator. Calcitonin secretion is stimulated by calcium and some gastrointestinal hormones (gastrin, cholecystokinin, and glucagon). The primary function of calcitonin is unclear as it appears to have a relatively minor role in calcium metabolism. It reduces serum calcium concentrations by direct inhibition of PTH and 1,25‐dihydroxyvitamin D‐mediated osteoclastic bone resorption. Calcitonin also increases the urinary excretion of calcium and phosphate but facilitates the absorption of nutrition‐derived calcium into blood. Bone has two main functions – (i) forming the rigid skeleton; and (ii) having a central role in calcium and phosphate homeostasis. Macroscopically, there are two types of bone: Bone consists of: Bone growth and reshaping take place throughout childhood and adolescence. Longitudinal growth of the long bones occurs by enchondral bone formation (Figure 10.2). In this process, cartilage cells (chondrocytes) proliferate in columns and undergo hypertrophic differentiation within the growth plate. Chondrocyte proliferation is regulated locally by the interaction of fibroblast growth factor with its receptor fibroblast growth factor receptor‐3 (FGFR3). Activating mutations of the FGFR3 are responsible for achondroplasia and hypochondroplasia. Hypertrophic differentiation of the chondrocyte is regulated by a negative feedback loop involving the cytokine parathyroid hormone‐related peptide (PTHrP) and a signalling molecule known as ‘Indian hedgehog’. The hypertrophic chondrocytes become surrounded by a matrix in which calcium is laid down. Newly calcified cartilage then condenses to become surrounded by osteoblasts which produce calcified osteoid. This calcified tissue is resorbed and replaced by bone trabeculae. By contrast, growth in width and thickness occurs by the process of intramembranous bone formation. This process occurs at the periosteal surface, without a cartilage matrix and with bone resorption taking place at the endosteal surface. Figure 10.2 Longitudinal growth of the long bones by enchondral bone formation. By contrast, bone remodelling is the lifelong process by which skeletal tissue is being continuously resorbed and replaced to maintain skeletal integrity, shape, and mass. In healthy individuals, the balance between bone formation and bone resorption is finely tuned. Bone mass increases progressively through childhood and adolescence until the maximum is attained in young adult life (so‐called ‘peak bone mass’) around 30 years old. Thereafter, a net loss of bone mass occurs as bone resorption exceeds the synthesis of new bone in later life. The amount of peak bone mass is a major risk factor for fractures in old age and is influenced by genetic factors, hormones, nutrition, and mechanical strain (Table 10.1). Table 10.1 Factors which influence peak bone mass. The causes of hypocalcaemia may be subdivided into those which present in infancy and those which present in older children, as shown in Table 10.2. PTH‐deficient hypoparathyroidism may be familial (autosomal dominant, recessive or X‐linked recessive), caused by activating mutations of the calcium‐sensing receptor gene or associated with other abnormalities, such as Addison’s disease or deafness. Non‐familial PTH‐deficient hypoparathyroidism (e.g. in association with DiGeorge’s syndrome) may present in infancy and be transient or persistent. In older children, it may be idiopathic, iatrogenic (e.g. following surgical removal of the parathyroids), or secondary (e.g. severe hypomagnesaemia). Table 10.2 Causes of hypocalcaemia. Pseudohypoparathyroidism is caused by resistance to the action of PTH. This is known as Albright’s hereditary osteodystrophy (AHO) when associated with the dysmorphic features described in Table 11.5 and Figure 11.4. The PTH resistance of AHO is most commonly associated with decreased G‐protein activity in cell membranes (type 1a), which may also affect other G‐protein coupled receptors (e.g. adrenocorticotrophic hormone (ACTH), thyroid‐stimulating hormone (TSH), luteinizing hormone (LH), follicle‐stimulating hormone (FSH) and glucagon). The genetics of AHO are complex, with parent‐of‐origin effects. Maternally inherited inactivating mutations of GNAS cause pseudohypoparathyroidism‐type 1a whereas paternally inherited mutations can lead either to pseudopseudohypoparathyroidism (the presence of clinical features of AHO but without biochemical evidence of PTH resistance) or to progressive osseous heteroplasia (ectopic bone formation within skin and muscle). Importantly, because of random monoallelic expression of the allele bearing the mutation that inactivates GNAS, any given hormone resistance may fluctuate over time, i.e. an infant with AHO may have normal calcium and develop severe hypocalcaemia later in life. The parent should therefore be made aware of the signs and symptoms that suggest hypocalcaemia. When assessing a child with hypocalcaemia, the following points should be highlighted in the clinical history: If an older child is suspected of having hypocalcaemia, latent tetany may be detected from positive Chvostek’s sign (facial twitching in response to tapping of the facial nerve in front of the ear) and Trousseau’s sign (carpal spasm within three minutes of inflating a blood pressure cuff above systolic blood pressure; Figure 10.3), or stridor. Extrapyramidal signs from basal ganglia calcification, cataracts, papilloedema, dry skin, coarse hair, brittle nails, and enamel hypoplasia with dental caries are signs suggestive of chronic hypocalcaemia. In contrast to the older child, hypocalcaemia in infancy is associated with relatively non‐specific symptoms including tremor, apnoea, cyanosis, and lethargy. Clinical signs suggestive of autoimmune hypoparathyroidism and pseudohypoparathyroidism are shown in Table 10.3 with details of the dysmorphic features of pseudohypoparathyroidism shown in Table 11.5 and Figure 11.4. Figure 10.3 Trousseau’s sign. Table 10.3 Clinical signs to look for on examination. If hypocalcaemia is suspected, a blood sample should be taken for the measurement of: The differential diagnosis of biochemical abnormalities of calcium metabolism is shown in Table 10.4. Table 10.4 Differential diagnosis of disorders of vitamin D and parathyroid hormone (PTH) metabolism. Source: From Ranke (1992). Ca2, calcium; PO4, phosphate; 25‐OHD, 25‐hydroxyvitamin D; 1,25‐(OH)2D, 1,25‐dihydroxyvitamin D; ↑, high; N, normal; ↓, low; LN, low–normal. Care must be taken when IV calcium is administered as extravasation of calcium into subcutaneous tissues around the injection site can lead to tissue necrosis; transfer to oral medication as soon as possible is advised. Long‐term maintenance treatment of hypocalcaemia requires vitamin D given as alfacalcidol (neonates 50–100 ng/kg daily, children aged 1 month to 11 years 25–50 ng/kg daily and children aged 12–17 years 1 μg daily) or calcitriol (children aged 1 month to 11 years initially 15 ng/kg daily, increased by 5 ng/kg daily to a maximum of 250 ng and children aged 12–17 years 250 ng daily increased by 5 ng/kg daily (maximum step 250 ng) every 2–4 weeks to a usual dose of 0.5–1.0 μg daily). These preparations are recommended because of their short half‐life and rapid cessation of action should toxicity occur. Vitamin D may need to be given in combination with calcium. Frequent monitoring of serum calcium concentrations is required shortly after starting treatment – weekly, if there is concern about the response to therapy. Once the patient has demonstrated a satisfactory response, serum calcium concentration and the urinary calcium:creatinine (UCa:Cr) ratio should be measured every three months thereafter, aiming to keep UCa:Cr <0.7mmol/mmol. Regular renal ultrasounds should also be performed to monitor for calcification. In children with hypoparathyroidism (particularly those due to mutations of the calcium‐sensing receptor gene), the aim of therapy is to achieve low normal serum calcium concentrations (2.0–2.25 mmol/L) [8.0–9.0 mg/dL] while avoiding urinary calcium:creatinine ratios greater than 0.7 mmol/L/mmol/L [0.16 mg/dL/mg/dL]. In those with pseudohypoparathyroidism, the resistance to PTH primarily affects the proximal renal tubule and not renal handling of calcium. Therefore, to avoid adverse bone sequelae, treatment should suppress PTH levels to the normal range, which may require maintenance of serum calcium concentrations towards the upper end of the normal range (2.25–2.5 mmol/L) [9.0–10.0 mg/dL]. In contrast to the management of primary hypoparathyroidism, there is less concern in pseudohypoparathyroidism about potential risks of adverse renal effects of hypercalciuria. Mechanisms for rickets: Rickets is caused by delayed matrix mineralization at the growth plate resulting in excessive accumulation of uncalcified cartilage and bone (osteoid) matrix. The most common causes of rickets are those associated with vitamin D deficiency. They present most frequently at times of rapid growth which may occur in infancy or in puberty, particularly in children with increased skin pigmentation. Vitamin D deficiency may be a result of dietary insufficiency, malabsorption (e.g. coeliac disease) or inadequate exposure to sunlight. There is a need for a greater clinical awareness of vitamin D‐deficient rickets in Western industrialized societies as the current prevalence, particularly in infancy and adolescence, seems higher than is often appreciated even in relatively sunny countries. To prevent rickets, it is recommended that food be supplemented with 400 units (10 μg) of vitamin D daily for all children aged under four years and adolescents, particularly girls with darker skins. Children who consume more than 500 ml of infant formula milk do not need vitamin D supplementation as the milk is already fortified. Rickets of prematurity is thought to be caused by calcium and/or phosphate deficiency rather than vitamin D deficiency. Hypophosphataemic rickets is an X‐linked dominant disorder which causes a failure of phosphate resorption in the proximal renal tubule. It is usually due to an inactivating mutation in the PHEX gene, which results in abnormal PHEX‐mediated up‐regulation of FGF23 activity. Vitamin D‐dependent rickets is very rare and is caused either by deficiency of 1a‐hydroxylation of 25‐hydroxyvitamin D (type 1) or resistance to 1,25‐dihydroxyvitamin D (type 2). The classification of the different forms of rickets and their biochemical characteristics are shown in Table 10.4. In severe renal failure, deficiency of 1α‐hydroxylase causes impaired 1,25‐dihydroxyvitamin D synthesis which, in conjunction with increasing serum phosphate concentrations, leads to hypocalcaemia and secondary hyperparathyroidism with bone disease (renal osteodystrophy). Both rickets and osteomalacia (defective mineralization of osteoid tissue) are common in the many causes of Fanconi’s syndrome and type 2 renal tubular acidosis. The metabolic bone disease is caused by a combination of phosphaturia‐induced hypophosphataemia, hypercalciuria, abnormal vitamin D metabolism, and renal insufficiency. If a child presents with rickets, then the following details should be elicited: The child should undergo careful clinical examination, including documentation of growth and pubertal status. The clinical signs to look for on examination are shown in Table 10.3. Rickets causes abnormal bone mineralization which may be evident on X‐ray. Additional radiological features commonly include a characteristic widening of the growth plate with cupping, splaying and fraying of an irregularly margined metaphysis (Figure 10.4). The characteristic clinical signs of vitamin D‐deficient and hypophosphataemic rickets are detailed in Table 10.3 and Figure 10.5 and Figure 10.6. Figure 10.4 X‐ray of the wrist of a child with rickets caused by vitamin D deficiency. Figure 10.5 Rachitic rosary. Source: Courtesy of Dr Nina Ma. Figure 10.6 Bowing of the legs in a child with hypophosphataemic rickets. The lowered renal threshold for resorption of phosphate, which is associated with hypophosphataemic rickets, can be calculated from simultaneous serum and urinary biochemical measurements by reference to Kruse et al. (1982) or Shaw et al. (1990), which also contains details of age‐appropriate reference ranges. The presence of glycosuria, amino‐aciduria, or a metabolic acidosis with inappropriately alkaline urine is suggestive of a wider defect of tubular function, such as Fanconi’s syndrome or renal tubular acidosis. Depending on the cause of the rickets, different preparations of vitamin D are required for effective treatment, as shown in Table 10.5. Where renal dysfunction is associated with metabolic bone disease, treatment of the wider systemic metabolic abnormality is required. Detailed guidance for this is beyond the scope of this volume and the reader is advised to consult appropriate textbooks on paediatric nephrology. Children with Fanconi’s syndrome may require phosphate therapy, whereas those with renal tubular acidosis require treatment with bicarbonate. Table 10.5 Treatment of rickets. Unlike in adults, osteoporosis in childhood is not clearly defined but is caused by reduced bone mass per unit volume with a normal ratio of mineral to matrix. The possibility of osteoporosis should be considered in a child who presents with fractures following minimal trauma in whom there is also evidence of decreased bone mineral density (usually more than three standard deviations below the mean for age, size, and puberty). This may occur as a result of an imbalance between bone formation and bone resorption because of a variety of reasons or as a result of abnormalities of type 1 collagen synthesis (e.g. osteogenesis imperfecta). The causes of decreased bone mineral density, which may predispose to osteoporosis and present in childhood, are shown in Table 10.6. Table 10.6 Causes of osteoporosis and other disorders which may lead to decreased bone mineral density. Osteoporosis may present with obvious signs of a long bone fracture following minimal trauma or with back pain and deformity because of underlying vertebral compression fractures (Figure 10.7). Clinical assessment and investigations should aim to exclude the causes of decreased bone mineral density shown in Table 10.6. If a child presents with osteopenia, then the following details should be elicited: Figure 10.7 X‐rays showing vertebral compression in a child with osteoporosis. Clinical examination should include a careful assessment of growth, nutritional state, pubertal development, and the clinical signs of osteogenesis imperfecta listed in Table 10.3. The presence of unexplained bone tenderness and pain, particularly in the spinal region, and in association with a spinal deformity, such as a kyphosis or scoliosis, might suggest previously unsuspected fractures. These may occur with severe osteoporosis or osteogenesis imperfecta. Distinguishing between osteogenesis imperfecta and idiopathic juvenile osteoporosis may prove difficult. Osteogenesis imperfecta should be considered in the presence of the clinical signs listed in Table 10.3. A skull X‐ray for the presence of wormian bones and a blood sample for genetic testing (e.g. the type 1 collagen genes COL1A1 or COL1A2) may assist in the diagnosis of osteogenesis imperfecta (for classification, see Table 10.7). Idiopathic juvenile osteoporosis may be suggested by X‐ray evidence of metaphyseal compression fractures at the knee. Histomorphometric examination of bone biopsy material may also help distinguish between these two conditions. Table 10.7 Classification of osteogenesis imperfecta. Bone mineral density may be measured by dual energy X‐ray absorptiometry (DEXA) scanning of the lumbar spine or hip. This method involves minimal radiation exposure and is based on the measurement of the attenuation of X‐rays of differing energy intensity as they pass through bone. However, these characteristics are influenced by the width of bone through which the X‐rays pass, which in children may vary as a consequence of age, body size, or puberty, in addition to illness. Interpretation of the data needs to take these factors into account and appropriate paediatric reference values must be used to interpret the results. It is recommended that bone mineral density should only be measured in children with risk factors for low bone density associated with low trauma or recurrent fractures, back pain, spinal deformity or loss of height, change in mobility status, or malnutrition. Newer techniques for obtaining volumetric measures of bone mineral density that are not constrained by the limitations of DEXA, such as quantitative computed tomography, are now becoming available. The following measures may help in the treatment of osteoporosis: Hypercalcaemia occurs when the serum calcium concentration exceeds 2.65 mmol/L. The causes of hypercalcaemia are shown in Table 10.8. Table 10.8 Causes of hypercalcaemia. Mild idiopathic hypercalcaemia of infancy presents with symptoms between the ages of two and nine months, but resolves spontaneously by the age of four years. When severe, it may present in the neonatal period in association with the dysmorphic features of Williams syndrome (Table 10.3 and Figure 10.8). Hypercalcaemia is associated with granulomatous disease (e.g. sarcoidosis and tuberculosis) as a result of the conversion of 25‐hydroxyvitamin D to 1,25‐dihydroxyvitamin D in granulomatous cells. Figure 10.8 Williams syndrome. Neonatal primary hyperparathyroidism is a serious disorder caused by homozygous mutations of the calcium‐sensing receptor gene, the product of which regulates PTH secretion. It causes anorexia, hypotonia, chest deformities, and respiratory distress. It is associated with a high mortality in the neonatal period and, to be successfully treated, requires urgent parathyroidectomy. Similar heterozygous mutations are responsible for autosomal dominant, familial hypocalciuric hypercalcaemia. Primary hyperparathyroidism as seen in association with pituitary adenomas and gastrinomas or other pancreatic tumours (multiple endocrine neoplasia type 1) or in association with medullary thyroid cancer and phaeochromocytoma (multiple endocrine neoplasia type 2A) is exceptional in the paediatric age group. These two groups of disorders are inherited in an autosomal dominant fashion and all offspring of affected individuals should be screened for these endocrinopathies. The presence of elevated serum concentrations of PTH will distinguish hyperparathyroidism from hypercalcaemia caused by vitamin D intoxication, idiopathic infantile hypercalcaemia, granulomatous disease, and malignancy. When assessing a child with possible hypercalcaemia, the following points should be highlighted in the clinical history: If hypercalcaemia is suspected, a blood sample should be taken for the measurement of: The differential diagnosis of biochemical abnormalities of calcium metabolism is shown in Table 10.4. The treatment of hypercalcaemia involves promoting a low‐calcium diet and where relevant, stopping vitamin D, calcium supplements and other drugs, such as thiazides, which increase calcium concentrations. Patients should be encouraged to drink plenty of fluids. If significant dehydration is present, this should be corrected with IV 0.9% saline. Prednisolone may be helpful if hypercalcaemia is secondary to sarcoidosis or vitamin D intoxication. If hypercalcaemia persists, bisphosphonate therapy may help by inhibiting mobilization of calcium from the skeleton. A parathyroid adenoma may be located by careful ultrasound examination and treated by surgical removal of the affected gland. Hyperparathyroidism caused by diffuse hyperplasia requires surgical removal of all four glands. Many causes of hypocalcaemia, rickets, and hypercalcaemia are transient and resolve in childhood without the need for referral onto adult services. However, where the underlying cause has potential implications into adult life (e.g. hypoparathyroidism or pseudohypoparathyroidism), then referral through local transition arrangements should be organized. Of note, even if well and currently asymptomatic, children known to be at risk of multiple endocrine neoplasia require referral to adult services for the organization of ongoing monitoring. Where there is an underlying genetic cause for a disorder of bone or calcium biochemistry which may have implications for future offspring, organization of genetic counselling may be particularly important. Given the importance of bone mineral accretion into the third decade of life, teenagers undergoing monitoring for osteopenia should also be referred. Discussion may need to take place between paediatric and adult specialists regarding the interpretation of successive bone mineral density scan reports, given that paediatric practice involves interpreting data for body size (using Z‐scores) whereas the tendency in adult practice is to interpret data relative to peak bone mass (T scores).
Calcium and Bone
Physiology
Introduction
Vitamin D
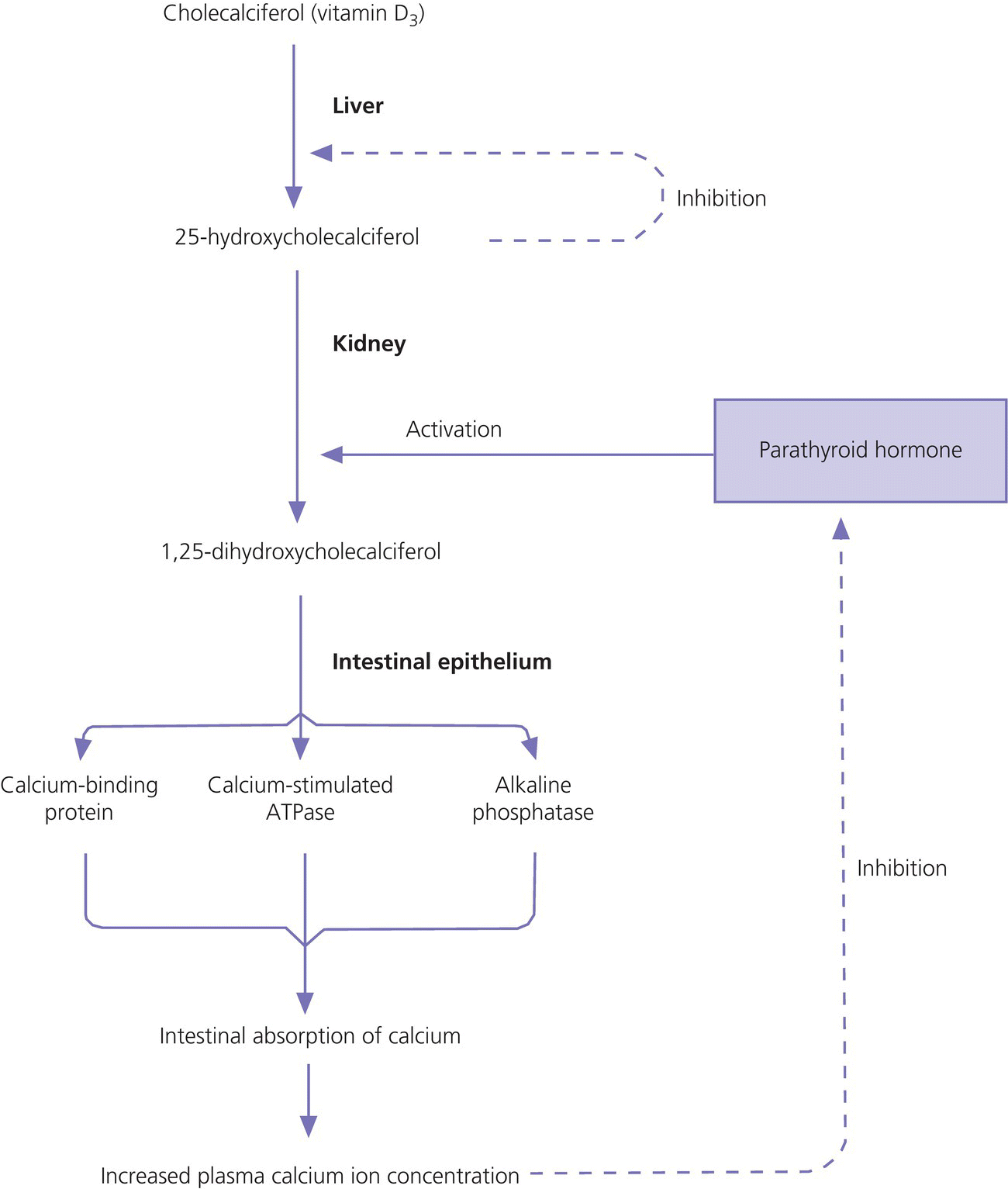
Parathyroid hormone
Calcitonin
Bone metabolism
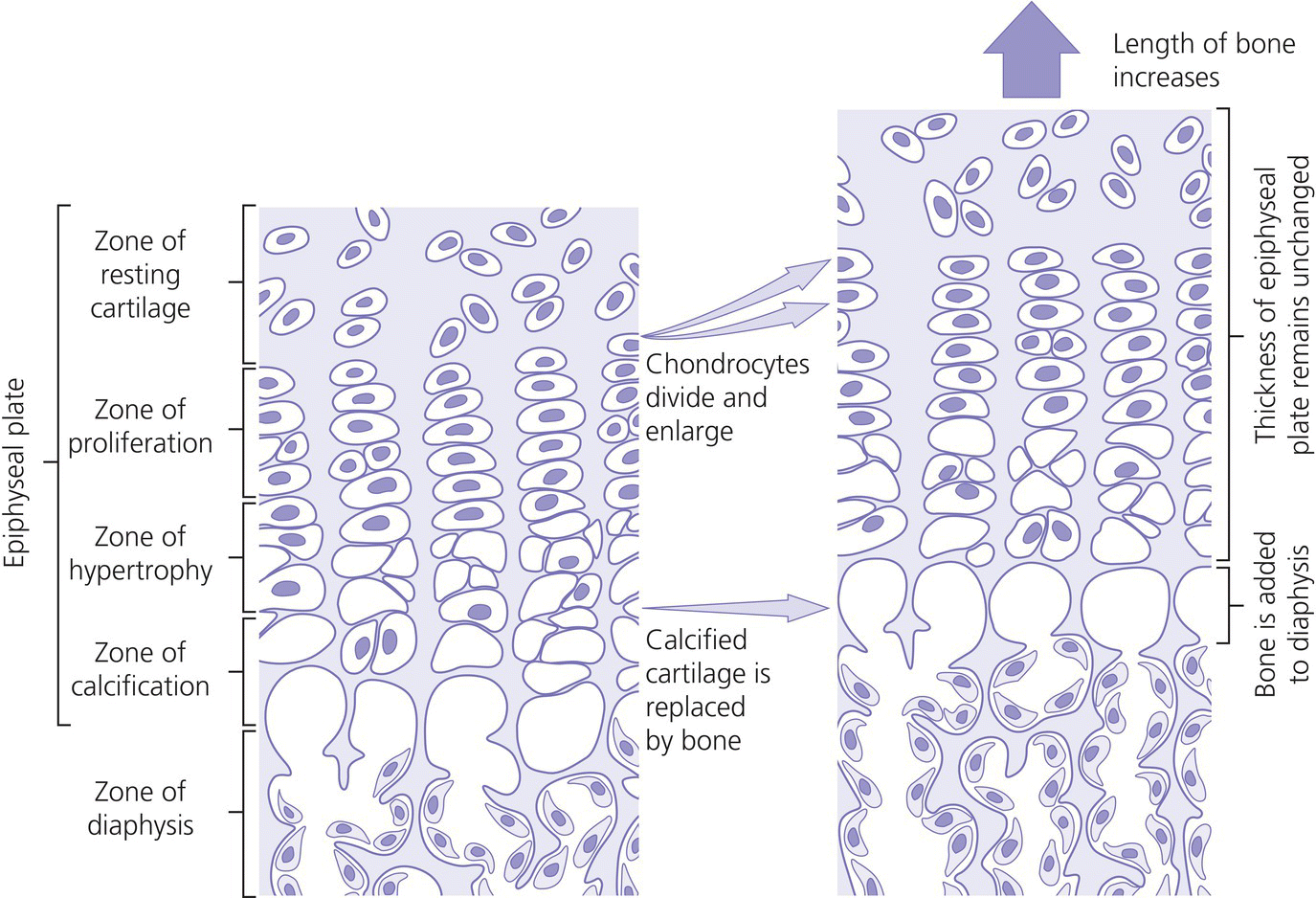
Influence
Increased bone mass
Decreased bone mass
Genetic
Afro‐Caribbeans
Caucasians
Orientals
Hormones
Calcitonin
Parathyroid hormone
Oestrogen
Vitamin D
Growth hormone
Glucocorticoids
Cytokines
Transforming growth factor β
Interleukin 1
Tumour necrosis factor α
Nutrition
Calcium
Anorexia
Obesity
Malabsorption
Mechanical
Exercise
Inactivity
Hypocalcaemia
Aetiology
Infancy
Childhood
Prematurity
Vitamin D deficiency
Asphyxia
Vitamin D‐dependent rickets (types 1 and 2)
Gestational diabetes
Chronic renal failure
Transient or permanent hypoparathyroidism
Hypoparathyroidism
Pseudohypoparathyroidism
High milk phosphate load
Hypomagnesaemia
Parenteral nutrition
Exchange transfusion
Chronic alkalosis or bicarbonate therapy
Maternal hyperparathyroidism
History and examination
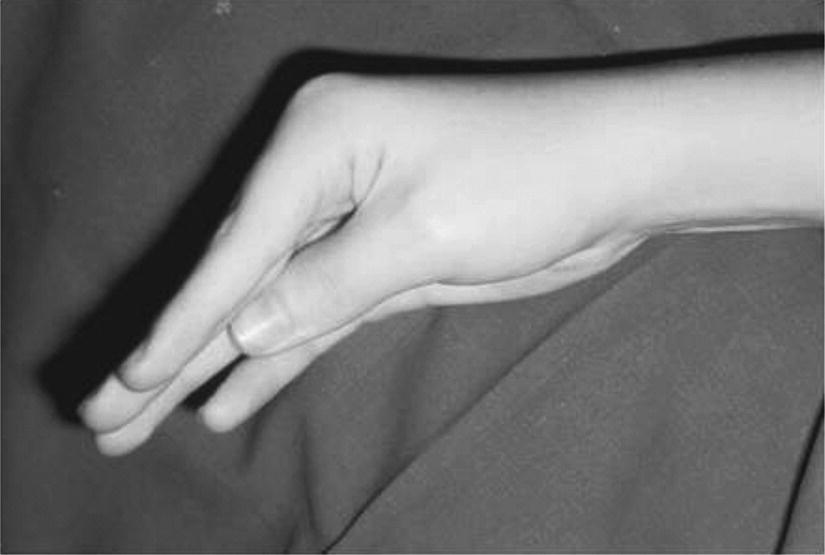
Clinical sign
Possible diagnosis
Candidiasis
Autoimmune hypoparathyroidism (polyglandular autoimmune disease type 1)
Dental enamel and nail dystrophy
Alopecia
Vitiligo
Signs of Addison’s disease or hypothyroidism
Short stature
Pseudohypoparathyroidism
Subcutaneous calcification
Dysmorphic features
Swollen wrists
Vitamin D‐deficient rickets
Prominent costochondral junctions (rachitic rosary) and Harrison’s sulci
Bow‐leg or knock‐knee
Craniotabes
Delayed dental eruption and enamel hypoplasia
Muscle weakness and tetany
Poor growth
Hypophosphataemic rickets
Bowing of the legs
Blue sclerae
Osteogenesis imperfecta
Abnormal dentition
Hyperextensible joints
Deafness
Broad and prominent forehead
Williams syndrome
Short turned‐up nose with flat nasal bridge
Overhanging upper lip
Late dental eruption
Supravalvular aortic stenosis
Peripheral pulmonary stenosis
Learning difficulties with emotional lability
Mild short stature
Alopecia
Vitamin D‐dependent rickets type 2
Investigations
Diagnosis
Ca2
PO4
PTH
25‐OHD
1,25‐(OH)2D
Vitamin D‐deficient rickets
LN
↓
↑
↓
↓, N, ↑
Vitamin D‐dependent rickets
Type 1 (deficiency of 1α‐hydroxylation)
↓
↓
↑
N, ↓
↓
Type 2 (resistance to 1,25‐(OH)2D)
↓
↓
↑
N, ↓
N, ↑
X‐linked hypophosphataemic rickets
N
↓
N
N
↓, N
Hypophosphataemic rickets with hypercalciuria
N
↓
N
N
↑
Tumour‐induced rickets
N
↓
N
N
↓
Renal osteodystrophy
N, ↓
↑
↑
N, L
Primary hyperparathyroidism
↑
↓
↑
N
N, ↑
Hypoparathyroidism
↓
↑
↓
N
↓, N
Pseudohypoparathyroidism
↓
↑
↑
N
↓, N
Vitamin D intoxication
↑
N
↓
↑
N, ↑
Hypercalcaemia in granulomatous disorders
↑
N
↓
N
↑
Treatment
Emergency management of acute hypocalcaemia complicated by tetany or a seizure
Rickets
Aetiology
History and examination
Investigations
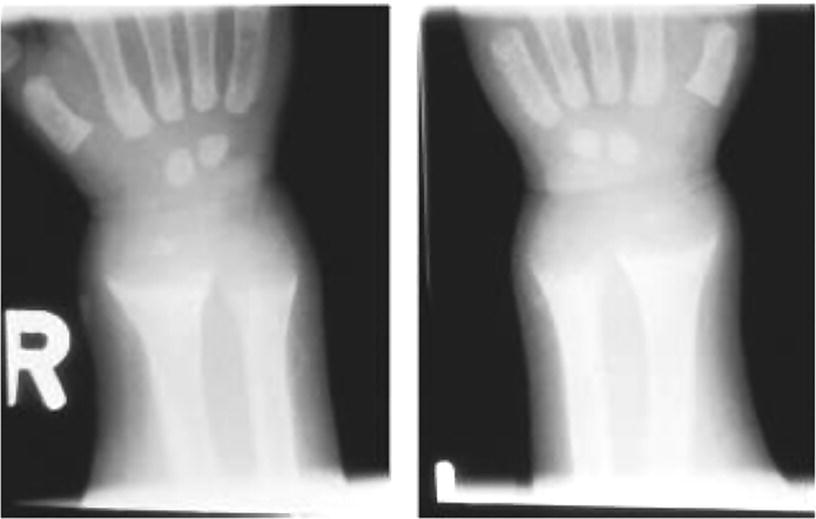
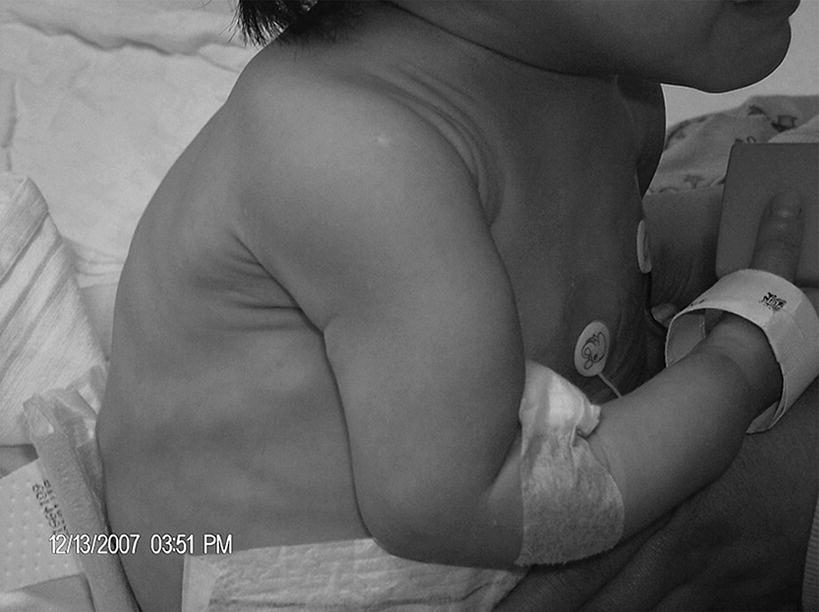
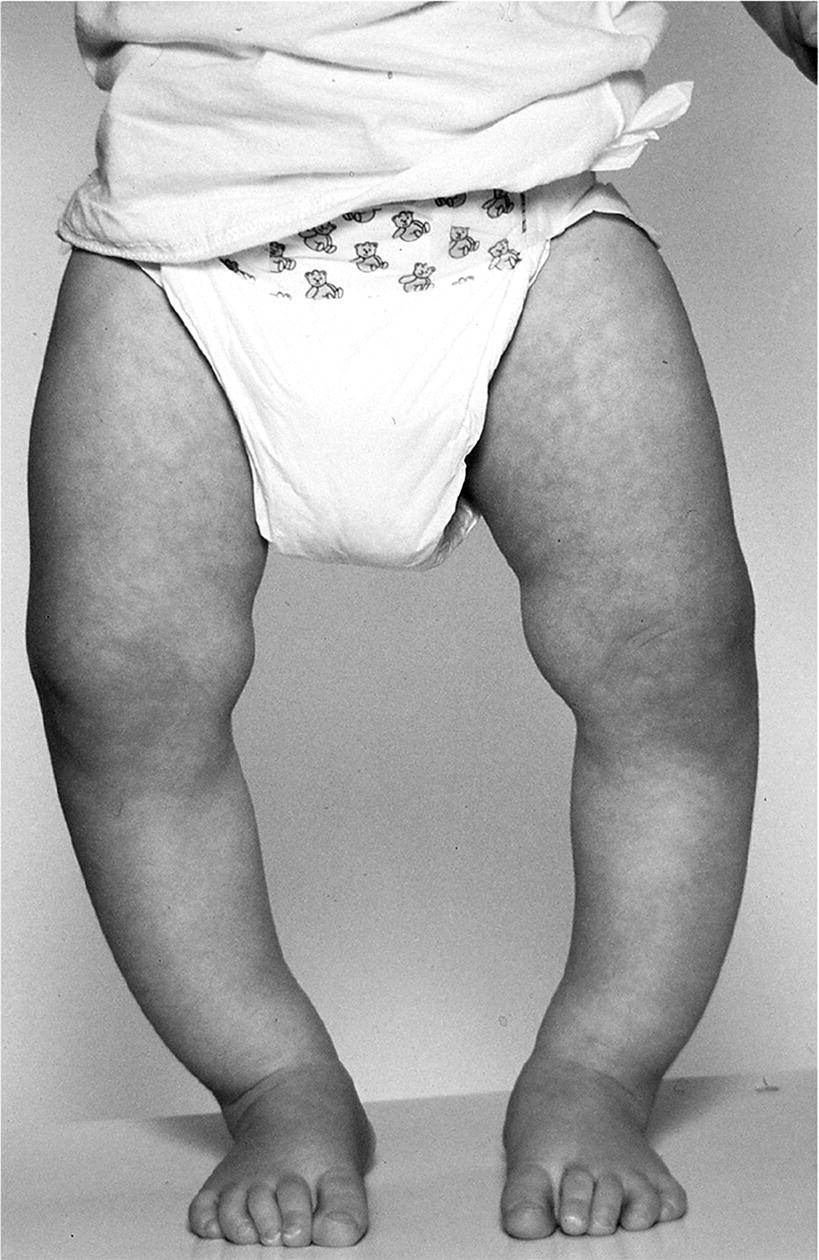
Treatment
Aetiology
Treatment
Vitamin D‐deficient rickets
Calciferol 150 000 units by intramuscular injection, repeated at 1–2 mo
OR
Cholecalciferol solution total dose of 150 000 units, preferably as a single dose orally or split over 3 d and repeated at 1–2 mo
Rickets of prematurity as a result of phosphate deficiency
Phosphate 1 mmol kg−1 daily in 1–2 divided doses by mouth or as an IV infusion
Vitamin D‐dependent rickets (type 1)
Calcitriol 0.25–2.0 μg per day
Vitamin D‐dependent rickets (type 2)
Calcitriol, large doses (up to 50 μg per day) and long‐term high‐dose oral or IV calcium
Hypophosphataemic rickets
Phosphate, 2–3 mmol/kg daily in 2–4 divided doses orally (maximum dose 48 mmol in children aged 1 mo–5 yr and 97 mmol in children aged 5–18 yr)
AND
Calcitriol 15–20 ng/kg per day increased to 30–60 ng/kg per day subdivided into one or two daily doses orally.
Osteoporosis
Aetiology
Pathogenetic basis
Examples
Primary bone disease
Idiopathic juvenile osteoporosis
Osteogenesis imperfecta
Osteoporosis pseudoglioma syndrome
Inflammatory
Inflammatory bowel disease
Rheumatoid arthritis
Endocrine
Cushing’s syndrome or disease
Drugs
Glucocorticoids
Others
Immobility
Acute lymphoblastic leukaemia
Thalassaemia
Homocystinuria
Post transplant
Causes of decreased bone mineral content (osteopenia) not necessarily severe enough to cause or osteoporosis
Anorexia, malnutrition
Coeliac disease
Hypopituitarism
Hypogonadism (e.g. Turner’s syndrome)
Hyperparathyroidism
Renal failure
Chronic liver disease
Thyrotoxicosis
Burns
Methotrexate
History and examination
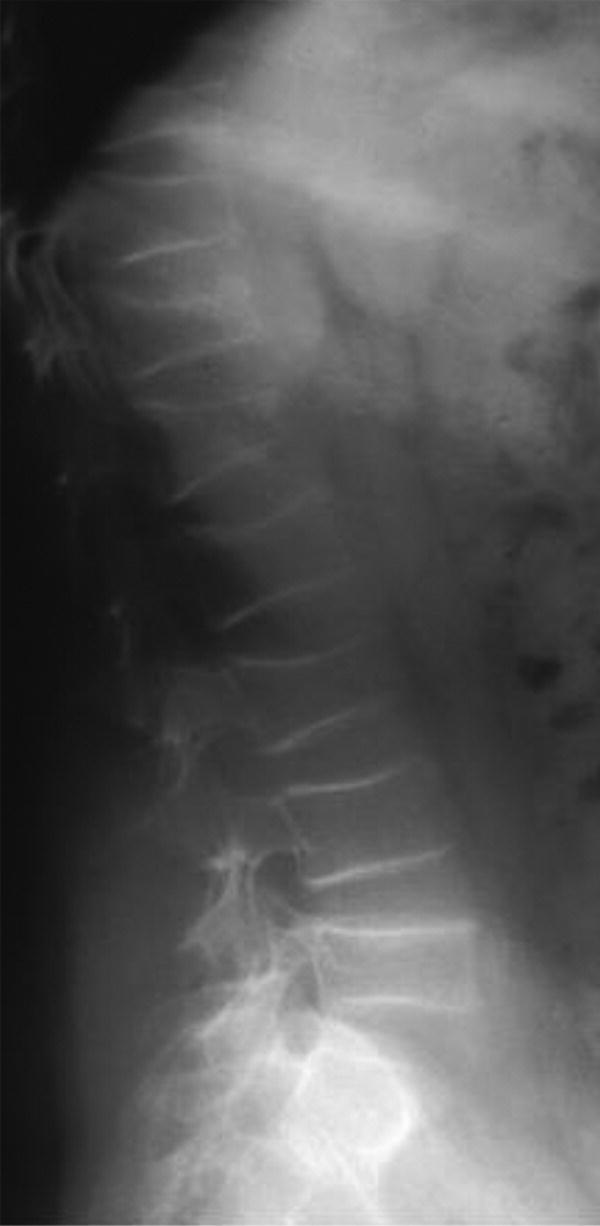
Type
Phenotype
Gene
Mode of inheritance
I
Mild, blue sclera, early deafness, mild stunting
COL1A1
Autosomal dominant
II
Severe, death in infancy
COL1A1, COL1A2
Autosomal dominant
III
Progressively deforming, infantile blue sclerae
COL1A1, COL1A2
Autosomal dominant
IV
Deforming, normal white sclerae
COL1A1, COL1A2
Autosomal dominant
V
Similar to IV but ‘mesh‐like’ bone
IFITM5
Autosomal dominant
VI
Similar to IV but ‘fish‐scale’ bone
SERPINF1
Autosomal recessive
VII
Severe to lethal
CRTAP
Autosomal recessive
VIII
Severe to lethal
LEPRE1
Autosomal recessive
IX
Severe
PPIB
Autosomal recessive
Investigations
Treatment
Hypercalcaemia
Aetiology
Neonatal or infantile onset
Gestational maternal hypocalcaemia
Idiopathic hypercalcaemia of infancy
Subcutaneous fat necrosis
Williams syndrome
Neonatal primary hyperparathyroidism
Preterm or intrauterine growth retarded‐associated phosphate depletion
Vitamin D intoxication (most commonly iatrogenic, e.g. during treatment of hypoparathyroidism)
Childhood onset
Parathyroid adenoma
Isolated familial hyperparathyroidism
Multiple endocrine neoplasia type 1
Multiple endocrine neoplasia type 2A
Familial hypocalciuric hypercalcaemia
Hypercalcaemia in granulomatous disorders
Acute lymphoblastic leukaemia
Immobilization
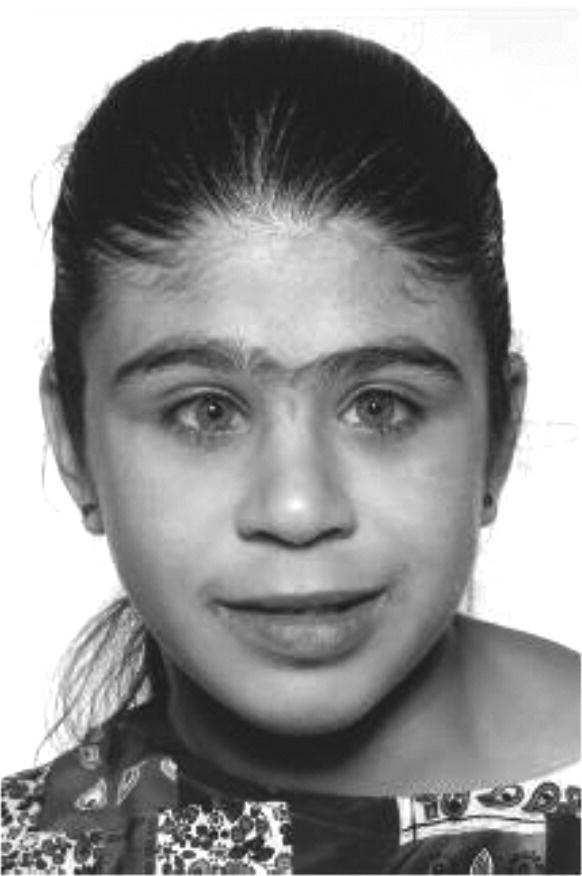
Neonatal hypercalcaemia
Hypercalcaemia in childhood
History and examination
Investigations
Treatment
When to involve a specialist centre
Transition
Future developments
Controversial points
Potential pitfalls
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


