Fig. 1
RET receptor activation. Shown is a schematic figure outlining the basic structural organization of the transmembrane receptor tyrosine kinase RET and the pathway to its activation. One of the four GDNF family ligands is recruited to the receptor upon binding to its cognate coreceptor. Solid arrows indicate the primary ligand/coreceptor interactions. Dotted arrows show secondary binding interactions. Generation of the heterotrimeric complex allows for receptor dimerization causing kinase activation and autophosphorylation (P). The predominant receptor complex observed in MTC cells is comprised of PSPN and GFRA4
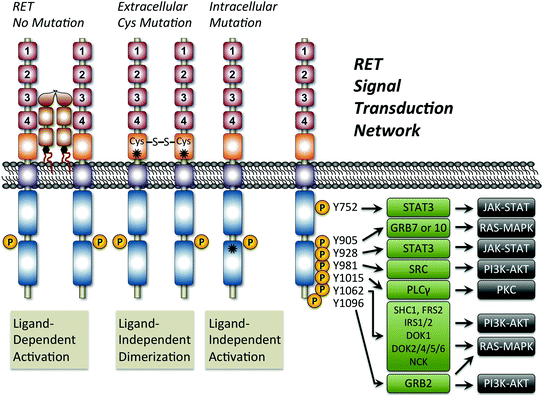
Fig. 2
Mechanism of oncogenic RET activation and the signal transduction network. Shown is a schematic figure outlining the normal ligand-dependent activation of RET compared with ligand-independent mechanisms resulting from receptor mutation. The asterisks show the general location of two common RET activating mutations. In all cases, RET activation is associated with phosphorylation (P) of tyrosines (Y) at seven possible positions located within the intracellular domain. Note that Y1096 only exists on the long isoform of RET. Tyrosine phosphorylation results in the recruitment of adaptor proteins (green boxes) that lead to the activation of multiple signaling pathways including JAK/STAT, PKC, PI3 K/AKT, and RAS/MAPK
6.1.1 Attenuated RET Signaling
RET signaling is crucial for mammalian embryonic development as mouse models lacking Ret function die before birth. The mutant embryos exhibit abnormal kidney development, and aberrant ureter, ovary, muscle, and intestine morphology. Ret signaling seems also essential for neurogenesis and establishment of neuronal populations of the central nervous system. In addition, Ret activation is necessary for the development of neural crest-derived lineages, in particular the enteric nervous system, adrenal medulla chromaffin cells and thyroid C-cells (Plaza-Menacho et al. 2014). Notably, loss of Ret, but not Gfra4 or Pspn, is associated with C-cell reductions in adult mice (Lindahl et al. 2000; Lindfors et al. 2006). Within the nervous system, GDNF/RET signaling is crucial to the maintenance of the nigrostriatal pathway by preventing cell death and onset of Parkinson’s disease (Kramer et al. 2007). It is also essential for the survival of motor neurons; alterations in the pattern of phosphorylation lead to amyotrophic lateral sclerosis (Luesma et al. 2014). RET signaling also maintains the enteric nervous system, and loss of RET function in these neurons leads to Hirschsprung’s disease, a condition characterized by the absence of enteric ganglia (Luesma et al. 2014). Importantly, this role of RET is reproduced in both Ret knockout (Schuchardt et al. 1994) and Ret knock-in mice with Ret C620R mutations (mixed Hirschsprung’s/MTC phenotype) (Carniti et al. 2006; Yin et al. 2007). Finally, it is important to remember that Gdnf/Ret signaling has a clearly established role in driving spermatogonial stem cell self-renewal in the testis and is therefore essential to male fertility (Meng et al. 2000; Hofmann 2008; Hofmann et al. 2005; Braydich-Stolle et al. 2007). Thus, in hereditary RETopathies or in therapies targeting RET activity, cell types beyond the thyroid C-cell must be considered.
6.1.2 Oncogenic RET Signaling
Several known mutations can convert the RET receptor into a dominant transforming oncogene, resulting in neoplasms affecting multiple cell types and organs. While RET mutation has been primarily associated with thyroid cancer, somatic structural RET alterations or changes of its expression have been described in lung, breast, and pancreatic cancers (Esseghir et al. 2007; Gainor and Shaw 2013; Ito et al. 2005; Kohno et al. 2012; Lipson et al. 2012; Morandi et al. 2013; Plaza-Menacho et al. 2010; Sawai et al. 2005). Germline activating mutations of RET are the cause of multiple endocrine neoplasia type 2 (MEN2), which is discussed in detail within other chapters of this book. Activating mutations of the RET oncogene fall into two major classes, extracellular and intracellular (Fig. 2). The extracellular cysteine-rich domain of RET is most frequently mutated at cysteine 634 (85 % of the cases), but mutations also occur distributed among all cysteines (C609, C611, C618, C620, C630) (Figlioli et al. 2013). Intracellular RET mutations are observed in the RET kinase domain and have a variable impact on RET activity. The most frequent mutation observed is M918T, which is found in patients with MEN2B and somatically in sporadic MTC. The molecular mechanism by which RET mutations trigger transformation has been elucidated (Santoro et al. 1995; Arighi et al. 2005; Santoro and Carlomagno 2013). As discussed, the normal route to activation involves binding of GFRA1-4/ligand complex to RET, which promotes dimerization and in turn autophosphorylation of the receptor. Mutations present in the extracellular domain are known to target highly conserved cysteine residues. Because these residues are normally involved in intramolecular disulfide bond formation, mutation generates a free sulfhydryl group. Mutant RET receptors in close proximity will then form intermolecular disulfide bonds, which function to stabilize dimerization even in the absence of the ligand (Fig. 2). This will therefore constitutively activate autophosphorylation and the intracellular pathways downstream of the receptor (Santoro et al. 1995; Asai et al. 1995; Borrello et al. 1995). In contrast, mutations in the intracellular tyrosine kinase domain of RET are known to target regions associated with the ATP-binding pocket, activation loop, and substrate binding pocket (Santoro et al. 1995; Salvatore et al. 2001; Songyang et al. 1995). Depending on their specific location, they induce structural changes and alter RET substrate specificity. This results in phosphorylation of alternative intracellular proteins. Therefore, the mutated receptor no longer needs dimerization to become active. RET mutations are often an initial step in the oncogenic process.
6.2 RAS Signaling
The RAS family of proteins consists of small GTPases that act as binary molecular switches downstream of ligand–receptor interactions (Malumbres and Pellicer 1998; Wennerberg et al. 2005). They are mainly activated through growth factors binding to RTKs, such as RET. RAS proteins are anchored to the plasma membrane through prenylation, which is indispensable to their function (Buss et al. 1989; Carboni et al. 1995; Cox et al. 1992). RTK autophosphorylation enables interaction with the adaptor protein GRB2 (Fig. 3). GRB2 is bound to SOS, a guanosine nucleotide exchange factor (GEF) that activates RAS by stimulating the release of guanosine diphosphate (GDP) to allow binding of guanosine triphosphate (GTP) (Cherfils and Zeghouf 2013). Therefore, upon stimulation by an activated receptor, RAS switches from the inactive “off” form (GDP bound) to the active “on” form (GTP-bound). Binding of GTP induces a conformational change leading to an increase in RAS affinity and binding to downstream mediators such as RAF or PI3K family members depending on the cell status. RAF is a serine–threonine kinase. RAS acts to recruit it to the plasma membrane and promotes dimerization and activation (Avruch et al. 2001). Activated RAF phosphorylates and activates the kinase MEK, which in turn phosphorylates and activates ERK kinase. Activated ERK phosphorylates a number of substrates, including other kinases and transcription factors, which will induce cell cycle progression (McCubrey et al. 2007). In the other main pathway, activation of PI3K will induce phosphorylation of AKT, leading to inhibition of several tumor suppressors (e.g., p21, BAD) or activation of the mTORC1 complex (Fig. 3) (Ma and Blenis 2009; Rodriguez-Viciana et al. 1994; Sarbassov et al. 2005; Staal 1987; Vanhaesebroeck et al. 2010) (Fig. 3). Note that activation of the PI3K/AKT pathway also occurs independently of RAS through direct and indirect binding of RTKs (Castellano and Downward 2011). The ultimate outcome is regulated induction of cell survival, growth, and migration. Components of these main signaling cascades are often mutated in cancer, with approximately one-third of human tumors expressing a constitutively activated mutant form of the RAS protein (Stephen et al. 2014). For a more extensive discussion of RAS signaling and targeting in cancer, the reader is referred to the following excellent recent reviews (Stephen et al. 2014; Prior et al. 2012; Cox et al. 2014; Pylayeva-Gupta et al. 2011; Malumbres and Barbacid 2003).
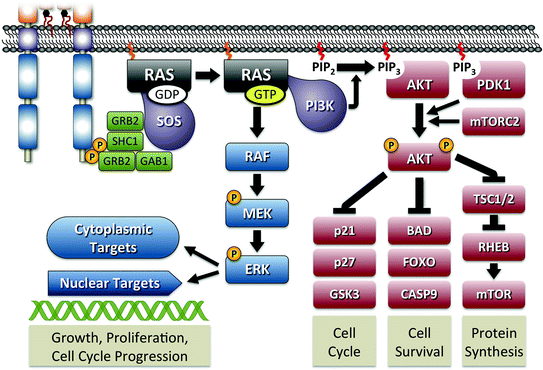
Fig. 3
RAS signaling pathway. The figure shows the activation of RAS initiated through interaction with phosphorylated RET. As shown in Fig. 2, signal transduction is primarily mediated by tyrosines 1062 and 1096 through adaptor proteins GRB2, SHC1, and GAB1. The guanine nucleotide exchange factor SOS is responsible for initiating the formation of active GTP-bound RAS. Active RAS is capable of activation of MAPK signaling through RAF or PI3K activation leading to AKT signaling. Major components of both pathways are shown with their specific outcome of regulating cellular functions indicated
6.2.1 Attenuated RAS Signaling
There are approximately 30 members of the RAS family, but the most clinically relevant are H-RAS, N-RAS, and K-RAS (Wennerberg et al. 2005). In mice, Kras knockout is embryonic lethal, whereas Hras, Nras, and Hras–Nras double knockouts survive normally (Esteban et al. 2001; Johnson et al. 1997; Koera et al. 1997; Umanoff et al. 1995). A thyroid C-cell phenotype has not been reported in any of these mouse models. Surprisingly, knockout of Nras, but not Hras or Kras, in combination with Rb1 haploinsufficiency is associated with a pronounced MTC phenotype in mice (see below). In human cell lines, somatic cell knockout of oncogenic KRAS has been accomplished in human colon cancer cells (Shirasawa et al. 1993). Upon KRAS gene disruption, these cells lost anchorage independence and grew slower than the parental cells in vitro and after injection into the flank of nude mice. They showed reduced expression of C–MYC, a transcription factor normally stabilized by phosphorylation in response to activation of the RAS/ERK pathway (Sears et al. 2000). To date, there have been no reports examining the impact of RAS loss in human MTC cell lines.
6.2.2 Activated and Oncogenic RAS Signaling
The RAS signal transduction pathway has been extensively studied in the context of tumorigenesis since the recognition that somatic gain-of-function mutations were responsible for cellular transformation. It is interesting to note that germline RAS gain-of-function mutations are observed in two separate genetic syndromes, both without a predominant cancer phenotype. Noonan syndrome, which is associated with heterozygous mutation of KRAS or NRAS, is characterized by learning disabilities, skin lesions, heart and facial abnormalities, cryptorchidism, and of potential relevance short stature with craniofacial anomalies (Cirstea et al. 2010; Schubbert et al. 2006). A role for the C-cell in any of these phenotypic features has not been reported. Costello syndrome is caused by heterozygous germline mutations at amino acid positions 12 and 13 in HRAS (Aoki et al. 2005). The result is a net increase in the active GTP-bound form of RAS. Interestingly, these positions are also the most frequently mutated positions in oncogenic RAS, and patients with Costello syndrome are at increased risk of neural crest tumors, although there have been no reports to date of MTC (Morandi et al. 2013; Gripp 2005). Those interested in RASopathies are referred to exhaustive reviews (Rauen 2013; Bezniakow et al. 2014).
Somatic gain-of-function mutations in RAS genes that promote cancer were identified more than three decades ago (for a review, refer to Malumbres and Barbacid 2003). Mutations are found in each of the three major isoforms and nearly all occur in the highly conserved codons 12, 13, and 61 (Prior et al. 2012; Cox et al. 2014). Mutation in these codons alters both nucleotide-binding properties and GTPase properties of the resulting RAS proteins. The primary outcome is a constitutive activation because RAS remains in the active, GTP-bound form (Milburn et al. 1990). Driver mutations involving RAS codons 12, 13, and 61 have been reported in a broad spectrum of cancer types, including MTC. The widespread involvement of RAS in tumorigenesis prompted the first MTC screen for somatic driver mutations in 1989 (Okazaki et al. 1989). The discovery of a single HRAS codon 61 mutation found in one of 18 tumors suggested that RAS mutation was a rare or low-frequency event. Three subsequent studies examining all three RAS family members in 30 MTC failed to uncover a single somatic mutation (Bockhorn et al. 2000; Horie et al. 1995; Moley et al. 1991). This lack of RAS involvement in C-cell oncogenesis fit well with in vitro studies performed during the same period, demonstrating that oncogenic HRAS and CRAF decreased proliferation through induction of neuroendocrine differentiation rather than enhancing tumorigenicity (Carson-Walter et al. 1998; Nakagawa et al. 1987). However, these results were in conflict with transgenic mouse model data, demonstrating that C-cells were sensitive to activated MAPK-induced tumorigenesis (Johnston et al. 1998; Schulz et al. 1992). More recently, the widespread availability of tools for somatic mutation analysis has led to a reexamination of RAS involvement in C-cell tumorigenesis. By focusing on identifying driver mutations in MTC tumors lacking somatic RET mutation, Moura et al. (2011) uncovered an unexpectedly high fraction of RAS mutation. Subsequent to these findings, several other studies have reported the presence of somatic RAS mutation (Agrawal et al. 2013; Boichard et al. 2012; Ciampi et al. 2013; Schulten et al. 2011). As seen in other tumor types, mutations are nearly exclusive to RAS codons 12, 13, and 61. The currently reported distribution of mutations seen in MTC differs from what is seen in other thyroid cancers with targeting of the HRAS gene, and specifically codon 61 mutations, occurring most frequently. In nearly all cases reported to date, RET and RAS mutations are mutually exclusive, supporting the belief that RET-mediated oncogenic transformation occurs separately from RAS.
6.3 Other Pathways
RET and RAS pathways have emerged as predominant oncogenic pathways for the C-cell; however, mutation of these genes currently only explains only about 60 % of MTC. The search for other major drivers of transformation has been largely unsuccessful. This failure may be explained in part by the lack of large unbiased search for new drivers. To date, only a single study reporting exomic sequencing was performed, which included only 25 MTC tumors (Agrawal et al. 2013). An alternative explanation is that a significant proportion of C-cell oncogenesis is driven by gene copy number changes rather than gene mutation. A role for copy number changes is supported by high-density comparative genomic hybridization (CGH) array studies (Flicker et al. 2012; Ye et al. 2008) and the unexpected MTC phenotype observed in numerous mouse models with allelic loss (see Table 1). From these studies, the retinoblastoma tumor suppressor (RB1) pathway has emerged as a route to C-cell oncogenesis in mice and should be considered a potentially important pathway in humans. Indeed, a recent association study specifically identified cell cycle genes in MTC development (Barbieri et al. 2014). The pathway primarily is made up of cyclin-dependent protein kinases (CDKs), cyclin-dependent kinase inhibitors (CDKNs), Cyclins (A, D, and E), RB proteins, and the E2F—family of transcription factors (Knudsen and Wang 2010) (Fig. 4). The pathway plays a critical role in cell cycle checkpoint regulation, which safeguards appropriate cell division; alterations in one or more of various components may lead to aberrant cell proliferation and development of cancer. Cell cycle progression is positively regulated by Cyclins activated by CDKs and negatively regulated by CDKNs. The activity of both is in turn regulated by environmental and cellular signals. For example, MAPK signaling leads to increased Cyclin D expression, while p53 activation enhances p21 function. The CDKN family members thus play a central role in preventing inappropriate cell division through negative CDK regulation and G1 cell cycle arrest. The CDKNs are made up of two major families—the INK4/CDKN2 family, which includes p15, p16, p18, and p19, and the Cip/Kip/CDKN1 family that includes p21, p27, and p57. The absence of CDKN suppression leads to increased phosphorylation of RB, which disrupts an inhibitory RB-E2F interaction. Once E2F is free of RB suppression, it functions to transcribe genes that are required for progression through S phase. The aberrant loss of CDKNs and RB has been associated with the development and progression of numerous cancer types and thus these proteins serve the role of tumor suppressors (Knudsen and Wang 2010; Zhu et al. 2014; Chinnam and Goodrich 2011; Di Fiore et al. 2013). Given the fundamental role that the RB1 gene played in the establishment of “two-hit” hypothesis of retinoblastoma (Knudson 1971), it was surprising that knockout of Rb1 in mice was initially associated with the development of pituitary tumors and subsequently CCH (Jacks et al. 1992; Williams et al. 1994; Harrison et al. 1995). Indeed, it is clear from several studies that Rb1 plays a central role in mouse C-cell tumorigenesis (see next section). Despite this observation, a direct role for RB1 loss in human C-cells has not been found. Studies that including RB1 sequence analysis failed to find mutation in 46 MTC tumors examined (Agrawal et al. 2013; Kanagal-Shamanna et al. 2014; Simbolo et al. 2014). A separate study found positive RB1 staining in 46 MTC tumors (Holm and Nesland 1994). When considered together, all these studies suggest that RB1 inactivation specifically induces C-cell oncogenesis but that mutation of the RB1 gene is a low-frequency event.
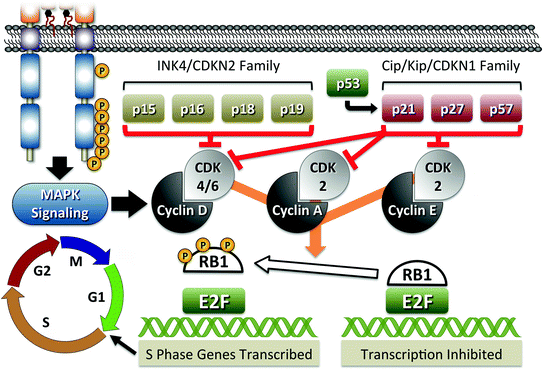
Table 1
Mouse models of MTC
Genetic target | C-cell phenotype | Reference |
|---|---|---|
RET | ||
Human RET C634R Tg-RET C634R short isoform Rat CT/CGRP promoter | MTC in 3 of 4 founder lines CCH or MTC in 93 % of mice MTC in 8, 13, and 14 months in each founder | Michiels et al. (1997) |
Human RET C634R Tg-RET C634R long isoform MoMuLV LTR promoter | Two founder lines only 1 with MTC Line 121 (22 copies) MTC in 100 % of mice by 9 months | Kawai et al. (2000) |
Human RET M918T Tg-RET M918T short isoform Human CT/CGRP promoter | MTC in 3 founder lines, highest in line 42 CCH in 77 % of line 42 mice by 8 months MTC in 13 % of line 42 mice by 11 months | Acton et al. (2000) |
MEN2B M919T knock-in retMEN2B /+ retMEN2B /MEN2B | Nodular CCH in 14 % at 8–12 months Nodular CCH in 60 % at 6–10 months | Smith-Hicks et al. (2000) |
MEN2A C620R knock-in retC620R /+ | No thyroid phenotype out to 24 months Homozygous is a neonatal lethal | Carniti et al. (2006) |
MEN2A C620R knock-in retC620R /+ | CCH in 48.4 % compared to 22.8 % in control mice at 20–30 months | Yin et al. (2007) |
Human RET C634R Tg-RET C634R long isoform Human CT/CGRP promoter CT-2A-3 (1 copy) | MTC in 0 FVB/n mice at 43 weeks MTC in 14 % of BALB/c mice at 43 weeks MTC in 64 % of C57BL/6J 9 mice at 43 weeks MTC in 98 % of CBA/ca mice at 43 weeks | |
Tg-RET M918T; p18+/+ No Tg-RET; p18+/− Tg-RET M918T; p18+/− No Tg-RET; p18−/− Tg-RET M918T; p18−/− No Tg-RET; p18+/−; p27+/− Tg-RET M918T; p18+/−; p27+/− No Tg-RET; p18−/−; p27+/− Tg-RET M918T; p18+/−; p27−/− | MTC in 0 % of mice by 12 months MTC in 0 % of mice by 12 months MTC in 26 % of mice by 12 months MTC in 11 % of mice by 12 months MTC in 43 % of mice by 12 months MTC in 0 % of mice by 12 months MTC in 27 % of mice by 12 months MTC in 88 % of mice by 9 months MTC in 100 % of mice by 9 months | van Veelen et al. (2008) |
RAS | ||
Tg-v-Ha-Ras Rat CT/CGRP promoter v-Ha-ras-p21 | MTC in 5 founder lines Expanded lines with 1 and 12 copies MTC in 85 % of mice at 6–12 months | Johnston et al. (1998) |
Other genes | ||
Tg-SV40 T-antigen Rat CT/CGRP promoter SV40 T-antigen | MTC in 5 founder lines MTC in 100 % of mice with death by 3 months | Baetscher et al. (1991) |
Tg-mos Moloney virus LTR Mouse mos | MTC in 3 of 4 founder lines MTC in 4–63 % of mice by 8 months | Schulz et al. (1992) |
Rb1+/−1t19neo | CCH in 43 % of mice | Harrison et al. (1995) |
Rb1+/− | MTC in 96 % of mice by 77 weeks | Zhou et al. (2005) |
Rb1 D326V/+ (M1326) Rb1 intron 10 3′ss/+ (M1033) Rb1 intron 21 5′ss/+ (M1032) | No thyroid tumors MTC in 90 % of mice by 70 weeks MTC in 90 % of mice by 70 weeks | Toki et al. (2014) |
Rb1+/− Rb1+/−; p53+/− Rb1+/−; p53−/− | MTC in 70 % of mice over 6 months MTC in 83 % of mice over 6 months MTC in 60 % of mice by 6 months | Williams et al. (1994) |
Rb1+/− Rb1+/−; p53+/− | MTC in 0 % of mice by 18 months MTC in 37 % of mice by 16 months | Harvey et al. (1995) |
Rb1+/−; p53+/− | CCH or MTC in 40 % of mice | Coxon et al. (1998) |
Rb1+/− p27−/− Rb1+/−; p27+/− Rb1+/−; p27−/− | MTC in 60 % of mice by 12 months MTC in 30 % of mice by 15 months MTC in 80 % of mice by 15 months MTC in 77 % of mice by 7 months | Park et al. (1999) |
Rb1+/−; Men1+/− | No effect on RB1 phenotype | Matoso et al. (2008) |
Rb1+/−; Msh2−/− | No effect on RB1 phenotype | Nikitin et al. (2002) |
p18−/− p27−/− p18−/−; p27+/− p18−/−; p27−/− | CCH 12 %, nodular CCH 2 % in mice over 12 months CCH 4 % of mice by 12 months CCH in 46 %, nodular CCH 8 % in mice by 9 months CCH in 81 %, nodular CCH 6 % in mice by 3.5 months | Franklin et al. (2000) |
p18−/−; men1+/− p27−/−; men1+/− | Nodular CCH in 80 % of mice by 22 months Nodular CCH in 6 % of mice by 12 months | Bai et al. (2007) |
p18−/− p18−/−; p53−/− | Nodular CCH in 2.4 % of mice at 14 months Nodular CCH in 5 % of mice at 8 months | Damo et al. (2005) |
Tg-TetOp-p25-GFP; Tg-NSE5021 | MTC in 100 % of mice by 16 weeks | Pozo et al. (2013) |
Rb1+/− Rb1+/−; E2f1+/− Rb+/−; E2f1−/− | MTC in 53 % of mice by 500 days MTC in 6 % of mice at 575 days MTC in 0 % of mice at 700 days | Yamasaki et al. (1998) |
Rb1+/− Rb1+/−; E2f3+/− Rb1+/−; E2f3−/− | MTC in 40 % of mice at 210–270 days MTC in 88 % of mice at 210–270 days MTC in 100 % of mice at 150–270 days | Ziebold et al. (2003) |
Rb1+/− Rb1+/−; Nras+/− Rb1+/−; Nras−/− | Nodular CCH or MTC in 32 % of mice MTC in 50 % of mice, significantly larger tumors MTC in 100 % of mice, significantly larger tumors | Takahashi et al. (2006) |
PRLR−/− | MTC in 29 % of mice at 12 months | Kedzia et al. (2005) |
Fig. 4
RB1-mediated pathway of cell cycle regulation. The figure shows a schematic representation of major proteins involved in RB1-mediated cell cycle regulation. RB1 is responsible for inactivation of E2F transcription factors involved in the expression of proteins required for G1 to S transition. Phosphorylation (P) of RB1 results in release from E2F and activation of transcription. RB1 phosphorylation is positively regulated through induction of Cyclins and CKDs, such as RET-mediated increase of Cyclin D, and negatively regulated through activation of CDK inhibitory proteins, the CDKN2 and CDKN1 families
In a similar manner, tumor modeling in mice has implicated roles for the RB1 regulatory CDKNs: p18 (CDKN2C), p21 (CDKN1A), and p27 (CDKN1B) in C-cell oncogenesis (see below). A role for CDKNs in MTC is supported by the observation that genetic loss of chromosome 1p (p18) is a frequent event in MTC (Mathew et al. 1987) and that reproducible losses targeting other CDKN genes are observed in recent CGH array studies (Flicker et al. 2012; Ye et al. 2008). Indeed, loss of the p18 region has been seen in 20 % of MTC tumors examined by CGH. A direct role for specific p18 loss in human MTC is also supported by a single study that found p18 mutations in 4 of 30 tumors examined (van Veelen et al. 2009). A caveat of these studies in that mutations were observed in patients with RET driver mutations, two of which were germline C634W mutations arguing against a driver role in human tumorigenesis. Additionally, an earlier study that included 50 MTC tumors found no p18 mutations (Holm and Nesland 1994), while a recent exomic study that included 17 tumors also found no mutations in any of the CDKN genes (Agrawal et al. 2013). The incongruity that exists between CGH and mutation studies suggests that as observed in mice, CDKN haploinsufficiency is sufficient to promote C-cell oncogenesis, or the inactivation of the remaining allele by mutation is a rare event. In support of a haploinsufficiency model, reduced p27 expression is significantly correlated with larger MTC tumor size and elevated serum calcitonin (Ito et al. 2005). Further investigation is needed to elucidate the role of the Rb pathway in MTC oncogenesis, a worthwhile endeavor given that targeted therapies, which function through direct CDK inhibition, are being developed to exploit these paths across many human cancers.
7 Modeling MTC in Animals
The first widespread animal models of MTC were the Long–Evan, Wag/Rij, and Sprague Dawley rat strains. The high incidence of spontaneous MTC tumors in these mice provided the tools for development of transplantable tumors and establishment of MTC cell lines (Muszynski et al. 1983; Zeytinoglu et al. 1980, 1983). However, despite the high frequency of MTC in these rat strains, the underlying genetic cause has only been identified in Sprague Dawley rats as mutation of the p27 gene (Cdkn1b) (Pellegata et al. 2006). For the Long–Evans and Wag/Rij rat strains, we can only say that germline mutations of the RET gene do not play a role (Fritz et al. 2002; De Miguel et al. 2003). With the development of tools to manipulate the mouse genome, rats were largely replaced as disease models for many disorders including MTC. This section focuses on engineered mouse models of MTC. A summary of mice with a reported MTC phenotype is provided in Table 1.
7.1 RET
Activating mutations within the RET gene were identified as a genetic cause of MTC in human in 1993 (Donis-Keller et al. 1993; Mulligan et al. 1993). With this discovery began the work to generate genetic mouse models to better understand the pathogenesis of the disease. Transgenic mice employing the CT/CGRP promoter for cell specificity predominate early models, which focused on the common RET mutations of C634R and M918T (Acton et al. 2000; Michiels et al. 1997; Reynolds et al. 2001). A single transgenic line has been created using nonspecific Moloney murine leukemia virus LTR (Kawai et al. 2000). A direct comparison of these models is difficult, given that in addition to the variable number of copies integrated, different transcription promoters were used along with expression of either long or short RET isoforms. Despite the variations, all mice were found to develop CCH as a precursor to MTC (Table 1). Curiously, unlike humans, the progression of MTC in mice expressing RET M918T is less aggressive than that observed in C634R models; this finding may relate to the use of the short RET isoform. Genetic knock-in models, which are clearly a more accurate representation of human disease, actually fail to develop MTC (Carniti et al. 2006; Yin et al. 2007; Smith-Hicks et al. 2000). However, similar to human disease, the M919T (in mice RET M919T is equivalent to the human MEN2B M918T) model develops earlier onset of CCH and pheochromocytoma compared to MEN2A models. Furthermore, MEN2A models display a pathology consistent with Hirschsprung’s disease, which is associated with the specific RET C620R mutation.
The differential onset of MTC in RET transgenic mice and failure to develop tumors in knock-in models argue for a role beyond aberrant RET activation in C-cell oncogenesis. Indeed, in comparing the models, the site of transgene integration may be more telling than expression levels. Two studies also support that RET-mediated oncogenesis requires a cooperative genetic background. First, Cranston et al. demonstrated that the development of MTC was highly strain specific. Using mice with a single RET C634R insertion driven from the CT/CGRP promoter, tumor production varied from zero in FVB/n mice to nearly 100 % in CBA/ca mice (Table 1) (Cranston and Ponder 2003). Of note, the RET MEN2B knock-in mouse model exists in an FVB/n mixed background. The second study is an extensive comparison of RET M918T-mediated tumorigenesis in mice with copy number variations of the cyclin-dependent kinase inhibitors p18 (cdkn2c) and p27 (cdkn1b) (van Veelen et al. 2008). This work demonstrates clear synergism between RET activation of MAPK pathways and deregulation of cell cycle progression (Table 1). These findings further correlate with the high frequency of allelic loss observed in chromosome 1p, where p18 maps, in human MTC tumors (Flicker et al. 2012; Ye et al. 2008) and the more aggressive phenotype observed in patients with coincident somatic M918T mutation and p18 loss (unpublished observations).
7.2 RAS
The first description of HRAS activation as an oncogenic driver of C-cells actually predates the discovery that RET germline mutations cause MEN2 (Okazaki et al. 1989). The development of a transgenic mouse with C-cell-specific expression of activated v-ha-ras occurred much later (Johnston et al. 1998). The v-ha-ras transgenic mouse most closely resembles the RET line generated by Michiels et al. (1997) in that both employ the rat CT/CGRP promoter and SV40 polyadenylation sites. Tumor formation in v-ha-ras mice is similar to that observed in RET transgenic mice (Table 1). Tumors were preceded by CCH, were calcitonin positive, and found to be invasive in older animals. Importantly, these findings are consistent with RET tumorigenesis being mediated through a ras-dependent pathway, but are in opposition to the neuroendocrine differentiation observed with ectopic expression in TT cells (Nakagawa et al. 1987). However, despite the high frequency of MTC formation observed in this model, a comparable genetic knock-in, hras G12V, has no overt tumor phenotype in animals up to 18 months of age (Chen et al. 2009; Schuhmacher et al. 2008). Furthermore, Costello and Noonan syndromes, which are associated with germline HRAS and KRAS/NRAS mutation respectively, have no reported incidence of MTC (Rauen 2013; Bezniakow et al. 2014). Therefore, a clear understanding of the role of RAS activation in C-cell oncogenesis remains to be elucidated.
7.3 Other Mouse Models with a MTC Phenotype
Three engineered mouse models with MTC actually predate the first reported RET transgenic mouse (Michiels et al. 1997). These include two transgenic models, one overexpressing c–Mos (Schulz et al. 1992) and the other SV40 T-antigen (Baetscher et al. 1991), and the earliest genetic knockouts of the Rb1 gene (Williams et al. 1994; Harrison et al. 1995; Harvey et al. 1995). Although the mechanism of Mos-mediated tumorigenesis was unclear, the authors speculated that Mos “may function in the same pathway that gives rise to MEN2.” The subsequent finding that MOS, like RET, activates the MAPK pathway serves to underscore the importance of MAPK activation in C-cell oncogenesis (Okazaki and Sagata 1995). For the remaining models, given that one of the actions of SV40 T-antigen is to inhibit Rb1 function (DeCaprio et al. 1988), these early findings served to identify regulation of cell cycle progression as a second key pathway of C-cell oncogenesis.
Since these initial studies, more than 20 engineered mouse models have been reported to have MTC as part of their phenotypic presentation (Table 1). These models nearly universally share aberrant upregulation of the MAPK pathway (Spry1) or targeted inhibition of RB1-mediated cell cycle control. A single exception is the observation that loss of prolactin receptor is associated with MTC formation (Kedzia et al. 2005). How loss of prolactin receptor causes C-cell transformation remains unclear though it is interesting that, like Rb1 loss, animals develop pituitary hyperplasia (Schuff et al. 2002). In looking at the remaining mouse models, some generalized observations seem to emerge. First, the targeting of p18 (Cdkn2c) or p27 (Cdkn1b), which indirectly inactivates Rb1 through enhanced phosphorylation, reproduces the Rb1+/− MTC phenotype albeit at a reduced level. Furthermore, as predicted, the simultaneous targeting of these Cdkn’s with Rb1 enhances C-cell oncogenicity; combination deletions with p53 or Men1, which act further upstream of Rb1, have only minor impact on tumorigenesis; and inactivation of E2f1, which is a downstream target of Rb1, inhibits tumorigenesis (Fig. 4, Table 1). Finally, targeted inactivation of Rb1 protein, mediated through its phosphorylation by hyperactive Cdk5, results in early onset of aggressive MTC with 100 % penetrance (Pozo et al. 2013). Despite the above observations, two additional Rb1 mouse models raise questions regarding its role in C-cell oncogenesis. Similar to the actions of Cdk5, Rb1 haploinsufficiency combined with either E2f3 or Nras loss results in the formation of highly penetrance aggressive MTC (Ziebold et al. 2003; Takahashi et al. 2006). The counterintuitive nature of these two observations serves to underscore the need for further investigation of pathways leading to C-cell oncogenesis.
8 Considering Mechanisms of C-Cell Tumor Initiation and Progression
Unlike some cancers, a clear stepwise route to C-cell oncogenesis has not emerged. Hereditary MTC demonstrates that activating mutations of RET serve as a primary initiating event. This role of RET is further supported by the observed high prevalence (~40 %) of RET mutations in sporadic MTC (Hu and Cote 2012). Several models have been proposed for how RET might function in initiation of C-cell oncogenesis. First, inactivation of RET in MTC cell culture models causes a marked reduction in tumor cell growth supporting a mechanism of oncogene addiction (Drosten and Putzer 2006; Vitagliano et al. 2011). This model is in part supported by patient responses to targeted therapies (Elisei et al. 2013; Wells et al. 2012). A second proposal is that RET increases genetic instability. In patient tumors, RET mutation is associated with increases in copy number variants and somatic mutations (Agrawal et al. 2013; Flicker et al. 2012; Ye et al. 2008). In one report examining sporadic MTC tumors using exomic sequencing, the mutation number was more than doubled in tumors with RET mutation (21.9 in RET+ vs. 8.4 in RET−) (Agrawal et al. 2013). In hereditary tumors, somatic mutation identified by exomic sequencing appears to be less frequent (Cai et al. 2015). Finally, a role for RET in altering the stem cell population needs to be considered in both hereditary and sporadic diseases. RET is known to play a central role in regulation of human spermatogonial stem cell self-renewal (Wu et al. 2009) and neural crest stem cell lineage derivation (Iwashita et al. 2003). In fact, somatic RET M918T mutation in male germ cells is associated with enhanced stem cell proliferation and provides a plausible mechanism for the paternal origin of MEN2B (Choi et al. 2012). While it remains to be formally examined, germline mutation of RET is also predicted to increase the neural crest progenitor cell population, thereby providing a larger pool of tumor initiating or the so-called cancer stem cells. A thyroid cancer stem cell model has been proposed for both medullary and nonmedullary thyroid cancers (Thomas et al. 2008; Zhang et al. 2006; Hardin et al. 2013). Support for a role in MTC derives from the observations that C-cells or their progenitors play a role in adult thyroid regeneration (Ozaki et al. 2012) and that RET mutation is associated with EGF/FGF-independent (Zhu et al. 2010), as well as 5-fluorouracil-resistant MTC tumorsphere growth (Kucerova et al. 2014).
Because MTC occurs in the absence of RET mutation, other pathways to C-cell oncogenesis must exist. Activation of RAS gene family members has emerged as a second oncogenic pathway with mutations observed in approximately 15 % of sporadic MTCs examined (Moura et al. 2011; Agrawal et al. 2013; Boichard et al. 2012; Ciampi et al. 2013; Schulten et al. 2011). A specific role for RAS as an initiator derives from the mouse models previously discussed, as patients with germline HRAS mutations (Costello syndrome) or KRAS/NRAS mutations (Noonan syndrome) have not been reported to develop MTC (Rauen 2013). Finally, no single gene has emerged as a common driver responsible for MTC without RET or RAS mutation. However, a focused examination remains to be performed. First, a recent comparison of Sanger-based sequencing with Ion Torrent AmpliSeq found a 30 % increase in the number of RET mutations detected (Simbolo et al. 2014). Furthermore, we have recently identified a case of RET gene fusion as a driver of tumorigenesis (Grubbs et al. 2015). RET fusions would go undetected by most DNA sequencing approaches. At the time of writing, only 5 such MTC tumors had been examined by exomic sequencing (Agrawal et al. 2013). The number of reported mutations in these 5 tumors ranged from 7 to 10, with only the TDG gene found mutated in more than one tumor. Unfortunately, Sanger sequencing could not confirm these specific TDG mutations, and TDG mutation was not in a separate larger validation set. While the small sample size may have prevented the discovery of other MTC genetic drivers, it is also possible that noncoding RNAs or gene copy number changes went undetected because of the limitations associated with the sequencing approach (Berindan-Neagoe et al. 2014; Yang and Lu 1839; Davoli et al. 2013; Manikandan et al. 2012). Indeed, in mice, heterozygous loss of Rb1 or p27 in the absence of a second mutation of the remaining allele is sufficient to induce MTC (Williams et al. 1994; Harvey et al. 1995; Park et al. 1999; Zhou et al. 2005). Although clear links establishing a role for cell cycle regulators in human C-cell oncogenesis do not exist, there is supportive evidence. Copy number changes that would not be detected by exomic sequencing have been observed using CGH arrays (Flicker et al. 2012; Ye et al. 2008). There have also been association studies linking increased MTC risk to genes associated with cell cycle regulation (Barbieri et al. 2014; Ito et al. 2005; Pasquali et al. 2011; Sekiya et al. 2014), although they are not universally positive (Sekiya et al. 2014). These observations fit well with a newer model, suggesting that heterozygous gene loss is sufficient to drive cancer (Davoli et al. 2013; Manikandan et al. 2012). The observation that MTC tumorigenesis in mice is significantly enhanced when targeting multiple Rb1 pathway members, particularly in combination with the RET/RAS pathway, suggests that progression and aggressive cancer might be caused by simultaneous hits to both pathways.
9 Summary
The production and secretion of calcitonin in response to increased serum calcium remains as the only well-defined role of the thyroid C-cell. The specific linking of the C-cell as the origin of MTC through calcitonin production has served as the primary motivator for understanding the biology of this cell. Ironically, this role has proven to be physiologically redundant, as genetic ablation of calcitonin is relatively inconsequential (Hoff et al. 2002). Unfortunately, little is known about other possible physiological roles of the C-cell and the mechanisms behind its unique sensitivity to RET-mediated oncogenesis. Genetic modeling in mice is beginning to gain insight into both processes. Studies of thyroidal development have suggested a possible interactive relationship between C-cells and follicular cells within the thyroid microenvironment (Andersson et al. 2011). Tumor modeling in mice has demonstrated that site-directed mutation of MEN2B-equivalent M919T alone is insufficient to cause MTC, but does induce pheochromocytoma. Models with unexpected MTC phenotypes have also linked oncogenesis to the Rb1-mediated pathway of cell cycle regulation. Together these pathways appear to synergize in a manner that appears to preferentially target the C-cell. Whether it is these cellular pathways that provide the C-cell with specific sensitivity to oncogenesis remains an unanswered question. Indeed, a role for RET signaling in normal C-cell function remains to be elucidated. In the nervous system, RET plays an essential role during development and in neuronal survival. A parallel role may exist for the C-cell, or perhaps like for the spermatogonial stem cell, RET functions as a C-cell signal for stem cell maintenance (Mulligan 2014). Roles for C-cells and calcitonin in thyroid regeneration and goiter formation have been proposed (Ozaki et al. 2012; Lupulescu 1972). Such a role would certainly fit with dysregulation of these pathways being associated with oncogenesis. Thus, while we have an ever-increasing knowledge base linked to the cellular drivers of C-cell oncogenesis, the primary role of these genes in normal C-cell physiology remains unclear. A greater understanding of the normal function of these genes is likely to contribute to defining the differences observed in the biology of MTC between mouse models and human, as well as offering greater insight into pathways of tumorigenesis beyond aberrant RET activation.
Acknowledgments
G.J. Cote is supported by NIH/NCI grant P50 CA168505.
References
Abu-Khudir R, Paquette J, Lefort A, Libert F, Chanoine JP, Vassart G, Deladoey J (2010) Transcriptome, methylome and genomic variations analysis of ectopic thyroid glands. PLoS ONE 5(10):e13420. doi:10.1371/journal.pone.0013420 PubMedCentralPubMed
Acton DS, Velthuyzen D, Lips CJ, Hoppener JW (2000) Multiple endocrine neoplasia type 2B mutation in human RET oncogene induces medullary thyroid carcinoma in transgenic mice. Oncogene 19(27):3121–3125. doi:10.1038/sj.onc.1203648 PubMed
Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, Bhan S, Ho AS, Khan Z, Bishop J, Westra WH, Wood LD, Hruban RH, Tufano RP, Robinson B, Dralle H, Toledo SP, Toledo RA, Morris LG, Ghossein RA, Fagin JA, Chan TA, Velculescu VE, Vogelstein B, Kinzler KW, Papadopoulos N, Nelkin BD, Ball DW (2013) Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab 98(2):E364–E369. doi:10.1210/jc.2012-2703 PubMedCentralPubMed
Ahlgren SC, Bronner-Fraser M (1999) Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Biol: CB 9(22):1304–1314PubMed
Andersson L, Westerlund J, Liang S, Carlsson T, Amendola E, Fagman H, Nilsson M (2011) Role of EphA4 receptor signaling in thyroid development: regulation of folliculogenesis and propagation of the C-cell lineage. Endocrinology 152(3):1154–1164. doi:10.1210/en.2010-0232 PubMed
Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, Matsubara Y (2005) Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 37(10):1038–1040. doi:10.1038/ng1641 PubMed
Arighi E, Borrello MG, Sariola H (2005) RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 16(4–5):441–467. doi:10.1016/j.cytogfr.2005.05.010 PubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


