BRCA1 and BRCA2
BRCA1 and
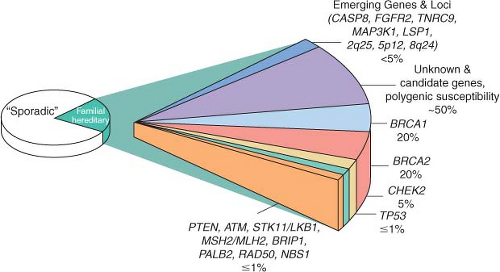
BRCA2 mutations account for approximately half of all dominantly inherited hereditary breast cancers. These mutations confer a relative risk of breast cancer 10 to 30 times that of women in the general population, resulting in a nearly 85% lifetime risk of breast cancer development.
5 BRCA1 and
BRCA2 mutation carriers are quite rare among the general population, however, the prevalence is substantially higher in certain founder populations, most notably in the Ashkenazi Jewish population, where the carrier frequency is 1 in 40.
More than a thousand germline mutations have been identified in
BRCA1 and
BRCA2. Pathogenic mutations most often result in truncated protein products, although mutations that interfere with protein function also exist.
4,
5 Interestingly, penetrance of pathogenic
BRCA1 and
BRCA2 mutations and age of cancer onset appear to vary both within and among family members. Specific BRCA mutations as well as gene-gene and gene-environment interactions as potential modifiers of
BRCA-related cancer risk are areas of active investigation.
6,
7BRCA1-related breast cancers are characterized by features that distinguish them from both
BRCA2-related and sporadic breast cancers.
4 BRCA1-related tumors typically occur in younger women and have more aggressive features, with high histologic grade, high proliferative rate, aneuploidy, and absence of estrogen and progesterone receptors and human epidermal growth factor receptor 2 (HER2). This “triple-negative” phenotype of
BRCA1-related breast cancers is further characterized by a “basal-like” gene expression profile of cytokeratins 5/6, 14, and 17, epidermal growth factor and P-cadherin.
8
Though
BRCA1 and
BRCA2 genes encode large proteins with multiple functions, they primarily act as classic tumor suppressor genes that function to maintain genomic stability by facilitating double-strand DNA repair through homologous
recombination.
8,
9 When loss of heterozygosity (LOH) occurs via loss, mutation, or silencing of the wild type
BRCA1 or
BRCA2 allele, the resultant defective DNA repair leads to rapid acquisition of additional mutations, particularly during DNA replication, and ultimately sets the stage for cancer development.
The integral role of BRCA1 and BRCA2 in double-strand DNA repair holds potential as a therapeutic target for
BRCA-related breast cancers. For example, platinum agents cause interstrand crosslinks, thereby blocking DNA replication and leading to stalled replication forks. Poly(adenosine diphosphate-ribose) polymerase-1 (PARP1) inhibitors additionally show promise as specific therapy for
BRCA-related tumors. PARP1 is a cellular enzyme that functions in single-strand DNA repair through base excision and represents a major alternative DNA repair pathway in the cell.
10,
11 When PARP inhibition is applied to a background deficient in double-strand DNA repair, as is the case in
BRCA-related tumor cells, the cells are left without adequate DNA repair mechanisms and ultimately undergo cell cycle arrest, chromosome instability, and cell death.
4 Given their phenotypic similarities to
BRCA1-related breast cancers, sporadic basal-like breast tumors may display sensitivity to PARP inhibition as well.
11 Phase 2 studies are currently under way to explore the use of PARP inhibitors in both
BRCA– and basallike, non-
BRCA-related breast tumors.
Other High-Penetrance Genes
A small number of other high-risk, low frequency breast cancer susceptibility genes exist, and they include
TP53, PTEN, STK11/LKB1, and
CDH1.4,
5,
6 These high-penetrance genes confer an eight- to tenfold increase in risk of breast cancer as compared to noncarriers, but they collectively account for less than 1% of cases of breast cancer. Like
BRCA1 and
BRCA2, these genes are inherited in an autosomal dominant fashion and function as tumor suppressors.
12 The hereditary cancer syndromes associated with each gene are usually characterized by multiple cancers in addition to breast cancer, as summarized in
Table 24.1.