The anatomy of the brain was described in Chapter 11. There are no significant anatomic differences between the central nervous system (CNS) of a child and the CNS of an adult.
The CNS in children reaches morphologic maturation during the first 2 years of life.
The brain’s neuronal complement and organization are essentially complete at birth; however, the myelin sheaths that cover the long nerve processes forming the connecting tracks or white matter of the brain and spinal cord are lacking. Myelinization occurs in a progressive anatomic sequence early in life, beginning with the corpus callosum centrally and ending with the white matter of the cerebral hemispheres peripherally at 12 to 24 months of age.
As functional maturation continues, the brain develops motor and sensory coordination during the first several years of childhood and progressive intellectual capacities throughout childhood and adolescence.
The type and degree of neurologic and neurocognitive alterations associated with brain irradiation correlate with age-related developmental status.
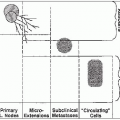
Medulloblastoma is an undifferentiated tumor believed to arise from the primitive multipotential medulloblast, embryologically located in the external granular layer of the cerebellum. It is classically identified as a primitive neuroectodermal tumor (PNET) presenting in the posterior fossa.
After much debate, the World Health Organization preserved the term medulloblastoma and identified the supratentorial PNET as a specific undifferentiated embryonal neoplasm separate from classic embryonal tumors, with clear lines of differentiation (Table 45-1).
The “staging” system is based on operative observation of tumor extent, now modified by imaging, and neuraxis staging, as suggested by Chang in the pre-computed tomography (CT) era (Table 45-2).
Current data indicate that M stage correlates significantly with outcome, but local tumor extent (T stage, including T3b or brainstem invasion) has little impact in series reporting aggressive surgical resection (1).
Risk stratification is performed following workup and resection and divides patients with medulloblastoma into either standard-risk or high-risk disease categories (Table 45-3).
The initial approach is gross total resection; complete or near-total resection is achieved in 70% to 90% of children and is associated with improved disease control (1).
Radiation therapy is often curative and is central in the management of medulloblastoma.
TABLE 45-1 Histopathologic Typing of CNS Tumors: World Health Organization Classification
Tumors of Neuroepithelial Tissue
Astrocytic tumors (astrocytoma, anaplastic astrocytoma, glioblastoma, pilocytic astrocytoma, pleomorphic xanthoastrocytoma, subependymal giant cell astrocytoma)
Oligodendroglial tumors (oligodendroglioma, anaplastic oligodendroglioma)
Ependymal tumors (ependymoma, anaplastic ependymoma, myxopapillary ependymoma)
Mixed gliomas (oligoastrocytoma, others)
Choroid plexus tumors
Neuronal tumors (gangliocytoma, ganglioglioma, desmoplastic infantile neuroepithelioma, dysembryoplastic neuroepithelial tumor)
Pineal tumors (pineocytoma, pineoblastoma)
Embryonal Tumors
Medulloepithelioma
Neuroblastoma
Ependymoblastoma
PNETs, medulloblastoma (posterior fossa, cerebellar), cerebral or spinal PNETs
Tumors of Meningothelial Cells
Meningioma
Malignant meningioma
Tumors of Uncertain Histogenesis
Hemangioblastoma
Germ Cell Tumors
Germinoma
Embryonal carcinoma Endodermal sinus tumor
Choriocarcinoma
Teratoma
Mixed germ cell tumors
Tumors of the Sellar Region
Pituitary adenoma
Craniopharyngioma
Note: Bold identifies embryonal tumors generically identified as primitive neuroectodermal tumors (PNET). Source: Modified from Kleihues P, Burger PC, Scheithauer BW, eds. Histological typing of tumours of the central nervous system. Berlin, Germany: Springer-Verlag, 1994.
Medulloblastoma is sensitive to chemotherapy; high response rates have been documented with alkylating agents (especiallycyclophosphamide) and platinum compounds (1).
The combination of aggressive chemotherapy with incomplete or inadequate craniospinal irradiation (CSI) is associated with high rates of neuraxis recurrence (1).
In patients receiving pre-CSI chemotherapy, the risk of neuroaxis progression increases with the duration of chemotherapy (21).
High-dose chemotherapy with autologous marrow rescue has occasionally been effective as a salvage regimen in recurrent disease (33).
TABLE 45-2 Chang Staging System for Medulloblastoma | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Because the entire subarachnoid space is at risk, full neuraxis irradiation is mandatory.
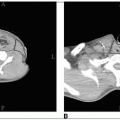
Adequate coverage at the subfrontal cribriform plate is particularly important; subfrontal recurrences are well documented and technically avoidable (Fig. 45-1) (19).
CSI is typically performed with the patient in a prone position by means of an immobilizing cast or vacuum device.
Lateral craniocervical fields adjoin a posterior spinal field (or two adjacent spinal fields in larger children).
The junction between the lateral and posterior fields is critical and is generally achieved with correction for both superior divergence of the posterior spinal field (using a collimator angle for
the lateral fields) and caudal divergence of the lateral fields (using a couch angle for the lateral fields). An “exact” three-dimensional (3-D) junction is preferable, obviating the need for a “gap,” yet requiring a shifting junction to minimize anatomically any dosimetric inhomogeneity (55).
TABLE 45-3 Medulloblastoma Risk Stratification
Standard (Average) Risk
>3 years old
<1.5 cm2 residual disease after resection
M0 by craniospinal MRI and CSF
High risk
<3 years old
Subtotal resection, >1.5 cm2 residual tumor
M+; leptomeningeal seeding
Location outside of posterior fossa (PNET)
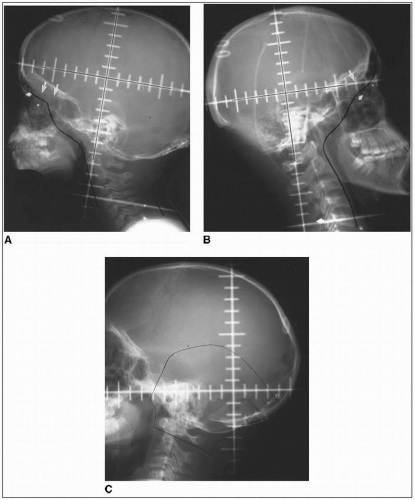
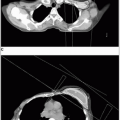
FIGURE 45-1 A: Craniospinal irradiation volume includes the entire intracranial subarachnoid space, covering the cribriform plate (arrows) in a 3-year-old child with close margin at the eye (markers). B: A wider margin is possible in a teenager with pneumatized frontal sinuses (arrows). C: Limits of posterior fossa fields include entire posterior fossa, based on tentorium (superiorly) in midsagittal MRI projection, posterior clinoid (anteriorly), and typically includes C1 to ensure coverage at the inferior margin of the posterior fossa. (Courtesy of M. Sontag, Ph.D.)
A frequently used “moving junction” allows for the elimination (or marked decrease) of dose inhomogeneity at the craniospinal portal junction and minimizes failures or complications (27).
The posterior orbit is not included for CNS tumors requiring CSI.
The spinal subarachnoid space extends caudally to S2 or beyond.
The lateral margins should dosimetrically include the width of the vertebral bodies beyond the neural foramina; a sacral “spade” is usually unnecessary, although in younger children the thoracic spinal component can be blocked to better spare the heart and lungs.
Use of electrons for spinal irradiation has been reported (35), but late results are unavailable. Electrons offer a potential advantage in limiting exit dose but require detailed attention to adequate coverage at depth and junctional homogeneity.
The posterior fossa “boost” is typically designed to encompass the entire infratentorial compartment (Fig. 45-1).
Investigations of 3-D conformal techniques and more localized boost volumes (using conventional, 3-D conformal, or stereotactic radiosurgical technique) are under evaluation (42).
The usual sequence of therapy is CSI followed by posterior fossa boost. Therapy may need to be initiated with posterior fossa irradiation if neurologic or hematologic status initially precludes accurate CSI.
Medulloblastoma is a relatively radiosensitive tumor. Local tumor control exceeds 80% with posterior fossa doses of 54 to 55 Gy (with field reduction after 45 Gy) in 1.6- to 1.8-Gy fractions (23).
With postoperative irradiation alone, the standard neuraxis dose is 35 to 36 Gy (1.5 to 1.8 Gy per fraction) (3).
A phase III trial of craniospinal radiation followed by chemotherapy demonstrated that average-risk patients may be treated safely with 23.4 Gy CSI (followed by a boost to posterior fossa to total 55.8 Gy) and concurrent vincristine followed by adjuvant chemotherapy (40). High-risk patients continue to require CSI doses of 36 Gy. The current COG standard-risk medulloblastoma trial is addressing further CSI dose reduction and boost volume reduction to an involved field versus the entire posterior fossa.
The World Health Organization classification identifies the specific histiotypes as medulloepithelioma, ependymoblastoma, or cerebral neuroblastoma; those without specific differentiation are classified as PNETs—medulloblastoma when located in the cerebellum, and PNET when supratentorial (Table 45-1).
Pineoblastomas are clinically grouped with the embryonal tumors.
Tumor extent and location often limit resectability of supratentorial PNETs, ependymoblastomas, pineoblastomas, and rhabdoid tumors.
Cerebral neuroblastomas are very often circumscribed lesions, with gross total resection reported in over 25% of cases (25).
Postoperative neuraxis irradiation with local boost to the primary tumor site is standard in children older than 3 to 4 years of age.
Guidelines for technique and dose are similar to those outlined for medulloblastoma: Boost volumes for supratentorial or pineal region tumors are defined as wide local volumes with 2- to 3-cm margins, based on preoperative tumor extent and postsurgical anatomic changes.
Limited data support local fields only for cerebral neuroblastoma; most series recommend CSI (31).
Ependymomas are derived from the ependymal cells lining the ventricular system; they occur throughout the CNS. They are seen infrequently in children.
In children, 90% of ependymomas are intracranial neoplasms; 60% to 70% arise in the posterior fossa, primarily within the fourth ventricle.
Supratentorial ependymomas occur predominantly in the parietal and frontal lobes, often contiguous with the ventricular system. They rarely occur as intraventricular tumors.
Posterior fossa lesions classically arise along the floor of the fourth ventricle and frequently extend through the foramen of Luschka toward or into the cerebellopontine angle.
In infants, tumors may originate in the cerebellopontine angle.
Tumors grow through the foramen magnum in up to 50% of cases, usually as tongue-like projections extending to the C1 or C2 level; caudal extension may reach to C5 (16).
In a review of 37 children, univariate analysis showed that total surgical resection and median infratentorial location correlated with better outcome (p < 0.002). Loss of differentiating structures or a combination of necrosis, endothelial proliferation, and mitotic index higher than 5 was associated with poor prognosis. Adjuvant chemotherapy or radiation therapy significantly enhanced progression-free survival only in patients who had incomplete tumor resection (9).
Maximal surgical resection is the optimal initial therapy (9), although it should possibly be delayed in infants in whom response to initial chemotherapy may permit more complete “secondary” resection.
For supratentorial tumors, size and location may limit resectability.
Radiation therapy adds to disease control and survival. Two retrospective reviews indicate survival of 0% and 13% with surgery alone compared with 45% and 59% with irradiation (p = 0.03) (44).
Ependymomas are relatively sensitive to chemotherapy, especially alkylating agents and platinum compounds.
A trial of adjuvant lomustine, vincristine sulfate, and prednisone showed no improvement in disease control (30).
Debate continues regarding the appropriate irradiation volume for intracranial ependymomas.
Some series indicate overt neuraxis dissemination at diagnosis in 3% to 16% of children; this is more often documented by cytology alone than by cranial and spinal imaging (44).
Although historic data suggested a correlation between high-grade or anaplastic posterior fossa ependymomas and spinal seeding, subsequent data failed to substantiate a site- or histology-specific relationship (3, 28, 44).
The incidence of neuraxis failure, either alone or in combination with local recurrence, is estimated to be 12% (29).
In most series, neuraxis failure is associated with simultaneous local recurrence in at least 50% of cases.
Disease control rates in contemporary series show no advantage to full cranial or craniospinal volumes compared with wide local fields, based on modern imaging (44).
The standard local volume for posterior fossa ependymomas encompasses the entire posterior fossa.
Fields are the same as those for medulloblastoma except in the lower margin; in ependymoma, the inferior limit is typically at the C2-3 interface.
For tumors extending into the upper cervical spine, the inferior margin should be two vertebral levels below the preoperative tumor extent until field reduction at 45 Gy.
Based on the recognized failure pattern at sites of identified invasion or disease residual, there now is greater acceptance of more localized treatment volumes. Prospective studies will address volumes defined by preoperative tumor extent (rather than the anatomically defined posterior fossa), with boost fields limited to sites of known invasion or residual disease (29).
For supratentorial ependymomas, wide local fields are defined by preoperative tumor extent, accounting for shifts in the normal brain postoperatively; margins of 2 to 3 cm are recommended (29).
For documented intraventricular extension, full ventricular irradiation (45 Gy) is appropriate.
Current guidelines call for 50 to 55 Gy to the primary tumor site, including field reduction at 45 Gy to more narrowly encompass the tumor bed (16).
Boost doses to 55 to 65 Gy have been recommended and are directed to small volumes of known residual disease, preferably using stereotactic radiosurgery or fractionated stereotactic irradiation (17).
Approximately 20% of pediatric brain tumors occur in infants and children younger than 3 years of age.
Compared with tumors in older children, those occurring in this age group are more likely to be malignant by histology (embryonal tumors, malignant gliomas, choroid plexus carcinomas, malignant rhabdoid tumors) and clinical behavior; supratentorial in location (especially during the first year of life); and associated with subarachnoid metastasis at diagnosis (44).
Surgery is more difficult in the infant brain.
The therapeutic index for radiation therapy is restrictive, with increased long-term neurologic and neurocognitive deficits leading to recommendations in dose reductions for children younger than 2 to 3 years of age (3).
Results for medulloblastoma, ependymoma, and pineoblastoma show less favorable prognosis for children younger than 3 to 5 years of age (3Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree