Fig. 8.1
Diabetes significantly affects osteoblast activity at multiple levels. Mesenchymal stem cells can differentiate into pre-osteoblasts or pre-adipocytes as well as other cell types. Runx2 is expressed when mesenchymal stem cells commit to osteogenesis and is required for osteoblast (bone formation cells) lineage selection. During the progression of osteoblast maturation stage, specific genes are expressed including osteocalcin, which is a late stage marker of osteoblast maturation. Following maturation, osteoblasts can undergo apoptosis or become embedded in the bone (osteocytes) during mineralization. Mesenchymal stems cells can also commit to adipogenesis. C/EBPβ promotes the expression of PPARγ2 which induces expression of other adipogenic genes such as aP2 (a protein that binds fatty acids). Osteoblast activity can be decreased by (1) promoting adipogenesis rather than osteogenesis, (2) inhibiting osteoblast maturation, and (3) increasing osteoblast apoptosis. Factors including TGF-β, Wnts, and BMPs increase osteogenesis, osteoblast maturation, and osteoblast viability. Diabetes can reduce osteoblast activity at all three levels of regulation. Factors such as diabetes-induced inflammation and reduced Wnt signaling can lead to suppressed osteoblast activity through modulation of the three levels of osteoblast regulation. These changes are thought to contribute to the decrease in bone formation seen in diabetes
Osteoblast activity is also regulated through modifying the rate/extent of cell maturation and death. During early stages of maturation, osteoblasts express collagen I, the major extracellular matrix component in bone [42]. Collagen I provides the foundation for subsequent matrix maturation and mineralization. During late stages of maturation, osteoblasts express osteocalcin, which is carboxylated at three residues to increase affinity to the bone matrix [43]. Not all osteocalcin is carboxylated. The undercarboxylated form can enter into the circulation to stimulate insulin secretion, insulin signaling sensitivity, and glucose homeostasis [43–46]; thus bone may affect and regulate diabetes. BMP and Wnt signaling promote osteoblast differentiation while many pro-inflammatory cytokines such as TNF-α reduce maturation. Once bone mineral is made, osteoblasts become osteocytes and embed within the mineralized bone, become bone lining cells that lie atop the newly formed bone, or undergo apoptosis [13, 21, 47, 48]. Increased osteoblast death is associated with decreased bone density due to the lack of bone forming cells. Metabolic stress (hypoxia, hyperglycemia) and pro-inflammatory cytokines can promote osteoblast apoptosis and bone loss [48–52].
Effect of T1 Diabetes on Osteoblasts
Decreased osteoblast activity is the key contributor to decreased bone mineral density observed in T1D children and adults [53–58]. Consistent with a decrease in bone mineral density, serum osteocalcin levels are typically lower in diabetic patients compared to nondiabetic subjects [46, 48, 53, 59–73] and are inversely proportional to glycosylated hemoglobin levels, a measure of metabolic control [74, 75]. Levels of serum undercaboxylated osteocalcin, which stimulates insulin secretion [59, 62, 76, 77], are negatively correlated with glycosylated hemoglobin levels in T1D male patients [78]. T1D rodents display bone loss similar to diabetic patients [68, 69, 71–73, 77, 79–82] and are useful models to study mechanisms of T1D bone loss. Specifically, diabetic mice and rats exhibit decreased trabecular bone volume and reduced cortical thickness (in some reports) within 4 weeks of T1D induction. T1D mouse and rat models also display decreased serum osteocalcin levels [64, 71, 73, 77, 80, 83] as well as decreased bone osteocalcin mRNA levels [46, 60, 62, 73, 84]. Histologic analyses indicate that osteoblast surface, mineral apposition and dynamic bone formation rates at trabecular, endosteal, and periosteal sites are reduced in T1D bone [52, 62, 76, 77, 85]. Bone implant integration and fracture healing are also suppressed in diabetic rodent models [72, 85, 86] consistent with an overall reduction in bone formation. Thus, a critical area of investigation is determining at what level (lineage selection, maturation, death) osteoblast activity is modified by T1D.
Lineage Selection
Several lines of evidence support that T1D alters MSC lineage selection. Most notably, mouse models of T1D display an increase in bone marrow adipocyte number and a decrease in the number of surface osteoblasts [14, 16, 18, 19, 45, 60, 87]. In vitro studies further indicate that factors present in T1D patients and mice (i.e., high glucose, pro-inflammatory cytokines) are capable of promoting MSC adipogenesis at the cost of osteogenesis [45, 49, 50, 60, 83, 88–90]. In addition, a recent study in rats found that the osteoprogenitor pool in T1D rat bone marrow was significantly depleted [88], further suggesting decreased lineage selection towards osteogenesis and/or a reduction in overall MSC number. Interestingly, the T1D adiposity is bone marrow specific, as T1D mice have depleted subcutaneous and visceral fat stores and exhibit weight loss [48, 83, 87]. This is consistent with the weight loss seen in T1D patients [87, 91]. Both T1D male and female mice display increased bone marrow adiposity in tibia, femur, and calvaria, and correspondingly display increased levels of adipocyte markers such as PPARγ2 and adipocyte protein 2 [aP2, also known as fatty acid binding protein 4 (FABP4)] in bone [45, 62]. T1D-induced marrow adiposity could be either an active or passive shunt of MSCs towards adipogenesis and away from osteogenesis [17–19, 92, 93] (see section on “Potential Mechanisms Contributing to Altered Bone Remodeling”).
Several in vivo studies have examined the role of T1D-induced marrow adiposity in mediating bone loss in T1D mouse models. Pharmacologically, an antagonist of PPARγ (bisphenol-A-diglycidyl ether) was given to T1D mice to inhibit adipogenesis. This approach was successful in preventing T1D-induced marrow adiposity, but T1D bone loss still occurred [23–25, 79]. Genetic manipulations, such as the knockout of C/EBP-β (a transcription factor required early in adipogenesis) have not been effective in preventing T1D bone loss [94, 95]. The lack of an association between bone marrow adiposity and bone loss was also observed in T1D patients [96], though marrow adiposity did correlate with serum lipid levels [96] suggesting a link to hyperlipidemia. Larger clinical studies are needed to determine significant relationships. Consistent with the lack of a link between marrow fat and T1D bone loss, mouse vertebral bone which has reduced density in T1D does not display an increase in marrow adiposity [45]. Taken together, these studies indicate that T1D is associated with changes in MSC lineage selection but T1D bone loss is not the direct result of increased marrow adiposity. Additional pathways regulating osteoblast activity must be involved.
Maturation
Once osteoblast lineage is selected, pre-osteoblasts progressively mature through stages of matrix production and mineralization. Identifying a specific change in maturation is somewhat difficult since altered lineage selection and increased death can also reduce markers of maturation in T1D bone. It is known that bone defect healing is decreased in T1D rats [97]. Similarly, T1D decreases bone formation on titanium and hydroxyapatite implants [80, 98, 99]. Though a reduction in pre-osteoblasts would cause similar outcomes, cell culture studies provide evidence to suggest that T1D reduces osteoblast maturation. Pre-osteoblast cell lines (i.e., MC3T3-E1 cells) cultured under conditions associated with T1D (high glucose or pro-inflammatory cytokines such as TNFα) display reduced levels of maturation markers [100–104]. In many of these conditions there is also evidence of enhanced expression of adipocyte markers (PPARγ and aP2) [45, 60, 62] suggesting a lineage or functional shift to an adipocyte.
Apoptosis
Increased osteoblast apoptosis can also contribute to reduced T1D osteoblast activity. T1D associated factors, such as reactive oxygen species (ROS), advanced glycation end-products (AGEs) and pro-inflammatory cytokines, are linked to increased osteoblast death [33, 48, 105–107]. In T1D mouse models, the number of osteoblasts stained by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), a method of detecting cellular apoptosis, was increased twofold compared to levels in control mouse femurs and tibias [52, 108]. The increase in apoptotic osteoblasts was confirmed in spontaneously diabetic Ins2 +/− mice where the increase was over threefold when compared to control [52]. Similarly, mRNA levels of Bax (a pro-apoptotic protein) were increased in streptozotocin (STZ)-induced diabetic mice and Ins2 +/− mice [52]. When osteoblasts (primary cells and cell lines) were cocultured with control versus T1D mouse bone marrow, the T1D marrow caused a twofold increase in osteoblast caspase-3 activity, a marker of cell apoptosis [52]. These findings support a role for the diabetic bone marrow microenvironment in inducing osteoblast death [52].
Effect of T2 Diabetes on Osteoblasts
In clinical studies, T2D patients display an increase, no change or decreased bone density [1–6]. Some studies found T2D to be associated with a decrease in serum osteocalcin levels in men and postmenopausal women [78, 109]. Variations in bone responses are likely a consequence of the complexity of the pathogenesis of T2D. Some T2D patients with above average weight and normal to high bone mineral density (factors typically associated with decreased risk fracture) also have increased risk fracture [110]. T2D can occur in non-obese and obese patients and thus bone changes can be associated with increased load, hyperlipidemia, hyperglycemia, inflammation and/or other complications that can contribute to altered bone density.
Accordingly, there are several T2D mouse models including obese and non-obese. In obese models such as T2D obese Wistar fatty rats, serum osteocalcin and bone osteocalcin levels are decreased as well as bone parameters such as length, strength, and weight [111]. Bone formation and turnover are both decreased in these T2D rodent models [112, 113]. In Zucker diabetic fatty (ZDF) rats (harbor a mutation in the leptin receptor, become obese rats and develop T2D at 9 week of age), a 55 % decrease in mineralized matrix formation was shown when bone marrow cells were stimulated by osteogenic media [114]. The same cells showed a 40–80 % reduction in Runx2, osteopontin, BMP-2, and osteocalcin mRNA expression. This data suggests that T2D may decrease lineage selection away from osteogenesis and decrease osteoblast maturation. The ob/ob mouse, a severe obesity model for T2D that harbors a mutation in the leptin gene, displays decreased amounts of trabecular and cortical bone and reduced femoral bone length, but increased trabecular bone volume in the lumbar vertebrae [115–119]. These mice also have decreased bone formation in both long bones and vertebrae [115, 119]. In the db/db T2D model (which harbor a mutation in the leptin receptor and become diabetic at 6–8 weeks of age) P. gingivalis-induced inflammation is exacerbated [112]. Bone formation was impaired and osteocalcin mRNA expression in the calvaria was decreased [112]. Caspase-3 and caspase-8 mRNA levels in the calvaria (apoptotic proteins) were increased over twofold when compared to control [83]. While osteoblast activity is decreased in these models, interpretations are complicated by not being able to distinguish bone response from T2D versus decreased leptin signaling.
Non-obese Goto-Kakizaki mice display reduced cortical bone thickness but this model showed abnormal pancreatic signs in the embryonic stage [33, 111]. Another non-obese T2D model, Torii rats, display suppressed osteoid surface, bone strength, and osteoblast numbers, which are all reversed by treatment with insulin [120]. A spontaneous T2D model (muscle IGF-1-1R-lysine-arginine, MKR) has a mutation in the skeletal muscle IGF-1 that leads to hyperglycemia by 7–8 weeks of age [110, 121–124]. MKR mice have decreased bone volume/total volume at the distal femoral metaphyses, stiffness, and failure load when compared to controls [122].
The Regulation of Osteoclasts and Bone Resorption by Diabetes
Regulation of Osteoclast Activity
Osteoclasts are multinucleated cells responsible for the resorption phase of the bone remodeling process. These cells are derived from HSCs, which reside in the bone marrow. Differentiation of HSCs into active osteoclasts is a multistep process (Fig. 8.2) and is regulated by three main cytokines: macrophage colony stimulating factor (M-CSF), RANKL and osteoprotegerin (OPG). These factors are derived from osteoblasts as well as other cells of the immune system including T-cells and B-cells [131]. While RANKL binds to RANK on osteoclasts to activate resorption, OPG (a secreted as well as membrane-bound protein produced by osteoblasts) acts as a decoy for RANKL, preventing it from binding to the transmembrane receptor RANK [137–139]. Both M-CSF and RANKL can increase nuclear factor-kappa (NF-kB) activity to stimulate osteoclast maturation [132–136]. RANKL does this by first inducing TNF receptor-associated factor (TRAF) proteins to start a signaling pathway that ends in the activation of NF-kB. NF-kB is typically bound in the cytoplasm by the inhibitor of kB (IkB). Binding of RANKL induces the phosphorylation of IkB to allow NF-kB to enter the nucleus to stimulate genes required for osteoclast function. Mutations in this signaling pathway lead to defective osteoclast formation and function [140–143]. Similarly, mutations in the gene encoding M-CSF causes osteopetrosis due to the inability of osteoclasts to fully mature.
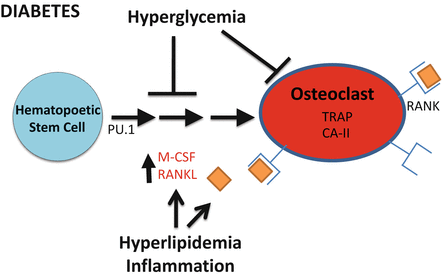
Fig. 8.2
Model for diabetes induced changes in osteoclast activity. Osteoclastogenesis involves early expression of PU.1 which regulates expression of the receptor for M-CSF. Early osteoclast maturation is induced by M-CSF and RANKL (expressed by osteoblasts). RANKL binds the RANK receptor on osteoclasts to activate NFkB activity, initiating the transcription of genes necessary for bone resorption. Diabetes-associated conditions such as inflammation (TNFα) and hyperlipidemia are known to activate osteoclast activity through increasing osteoclastogenesis and maturation. However, diabetes is typically associated with decreased bone resorption. This likely occurs due to other diabetes-associated factors such as hyperglycemia as well as expression of anti-inflammatory cytokines
Other factors involved in osteoclast differentiation include PU.1, which is a transcription factor that is required for the normal formation of osteoclasts. PU.1 regulates the receptor for macrophage colony stimulating actor (M-CSF) and integrin proteins needed for osteoclast maturation and binding of the osteoclast to the bone surface [125–127]. Mice lacking PU.1 die immediately after being born and have symptoms of osteopetrosis, an increase in bone density [128]. PU.1 interacts with other transcription factors (MITF, micro-ophthalmia-associated transcription factor) to regulate genes important for osteoclast function such as tartrate-resistant acid phosphatase (TRAP) and carbonic anhydrase 2 (CA-II) [129, 130].
Effect of T1 Diabetes on Osteoclasts
The majority of rodent studies examining T1D show no change or decreases in markers of osteoclastic resorption [33, 48, 62, 85, 144–147]. However, there is some controversy since a few studies have found increased osteoclast parameters in animals [147]. Clinical studies support no change in T1D osteoclast activity as determined by levels of serum markers of resorption such as deoxypyridinoline and c-terminal telopeptide of type 1 collagen (CTX) [53, 69, 86, 148]. In mice, the typical STZ dosing regimen used to induce T1D does not cause a change in osteoclast parameters (consistent with spontaneous mouse models [60]), however, increasing the dose causes an increase in osteoclast activity possibly due to more rapid onset of T1D and/or inflammation (as seen in the liver with high STZ dose) [67]. As noted previously, T1D increases bone marrow adiposity [60, 62] and thus MSC lineage selection. Not only does this change the overall physical morphology of the bone marrow compartment, but it also reduces the number of MSC maturing to the osteoblasts that are needed for HSC maintenance and maturation to osteoclasts (via M-CSF and RANKL) [149]; this can contribute to reduced osteoclastogenesis in the presence of inflammation. Other contributors to reduced resorption in T1D include hyperglycemia which can directly decrease RANKL-induced osteoclastogenesis [150], a reduced number of osteoblasts which can reduce RANKL levels in bone, and elevated levels of anti-inflammatory cytokines.
Effect of T2 Diabetes on Osteoclasts
T2D effects on patient bone densities are variable with reports indicating lower, the same, or higher bone density than nondiabetic individuals. In T2D animal models, such as the leptin receptor knock out db/db mouse, mice display suppression of bone remodeling, with both bone formation and resorption decreased [151]. Yet, osteoclast activity in T2D male patients has been reported to increase [152, 153]. Serum concentrations of TRAP and urine markers for bone resorption, such as CTX, deoxypyridinoline, and N-telopeptide of type I collagen (NTX), are significantly elevated in T2D patients [154–156]. However, T2D patients can also exhibit increased levels of OPG which can reduce the increased resorption caused by T2D [157].
Role of Marrow Immune Cells in the Regulation of Bone Remodeling in Diabetes
Bone Marrow: Bone Remodeling Signaling
Immune cells are capable of regulating osteoblast and osteoclast activity and vice versa. The role of immune cell populations and their effect on bone marrow hematopoiesis has been demonstrated in a variety of pathology conditions associated with bone loss. For example, activated T-cells are involved in regulating bone homeostasis in estrogen deficiency, an effect that was mediated by IFNγ [158]. In ovariectomized (ovx) mice, T-cells express high levels of TNF-α (enough to induce osteoclastogenesis) [159]. T-cell deficiency prevents ovx induced bone loss, a benefit lost when mice are given T-cells from WT mice but not T-cells from TNFα-deficient mice [160]. Similarly, blockade of TNF-α and IL-1 by Anakinra or Etanercept decreases bone resorption in postmenopausal women [161]. This suggests that an increase in these cytokines can be one of the causes of bone loss. However, TNF-α, IFNγ, and IL-1 are not the only cytokines associated with bone loss. Another cytokine IL-7 promotes osteoclastogenesis by upregulating T- and B-cell-derived RANKL [162]. IL-23 also stimulates the differentiation of human osteoclasts from peripheral blood mononuclear cells (PBMC) in the absence of osteoblasts or exogenous RANKL and in vivo blockade of IL-23 activity prevents inflammation and bone destruction in collagen-induced arthritis in rats [163]. These findings provide a new insight into the interaction between immune system, bone marrow and bone loss.
Another intracellular system that is associated with changes in the bone marrow, immune cells, and bone density is Wnt signaling. Wnt signaling regulates the reciprocal relationship between adipocytes and osteoblast. Part of the mechanism whereby Wnt10b inhibits adipogenesis and stimulates osteoblastogenesis is by suppression of PPAR-γ. In addition, it has been found that expression of Wnt10b in marrow increases trabecular bone mass and strength [37]. Wnt ligands are also required for hematopoietic progenitor cells (HSPC) expansion and survival. Experiments in ovx mice has shown that T-cell-expressed CD40 ligand (CD40L) enhance T-cell production of Wnt10b which results in activation of stromal cell (SCs) and HSPCs, along with improvement of cytokine production by SCs [164]. In summary, these results recognized the impact of cellular interaction between immune cells, HSPCs and the effect in bone homeostasis.
Effect of T1D on Marrow Cells
T1D is an autoimmune disease characterized by destruction of pancreatic β-cells producing insulin. White blood cells, like T-cells and macrophages, and the interaction and secretion of cytokines, nitric oxide, and free radicals are involved in this autoimmune pathology. Cytokines, such as IFNγ, IL-1β, and TNFα, can act together to induce beta cell death and can also affect the expression of genes that are protective or harmful for beta cell survival such as signal transducers and activators of transcription 1 (STAT-1) and interferon regulatory factor 1 (IRF-1) [169]. One study examining T-helper cell cytokine profiles indicated higher intracellular TNFα in CD8+T cells at the time of T1D diagnosis and higher intracellular TNFα in CD4+T lymphocytes in patients at 3 months post-diagnosis, compared with controls [170]. This suggests that there are distinct roles for cytokine release during different stages of T1D. Diabetic mouse models also display elevated TNFα levels in serum and increased TNFα mRNA level/expression in bone [52, 72]. In the STZ-induced diabetic mouse model IL-1α, IL-6, IFNγ, and TNFα expression are increased in bone with the onset of T1D and during the onset of altered expression of bone phenotype markers [72] (Fig. 8.3). Furthermore, it has been shown that T1D reduces the number of MSCs found at sites of fracture repair and that suppression of TNFα with Pegsunercept treatment restores the level of MSCs to control levels. It was further determined that TNFα is capable of reducing MSC proliferation and increasing MSC apoptosis [171].
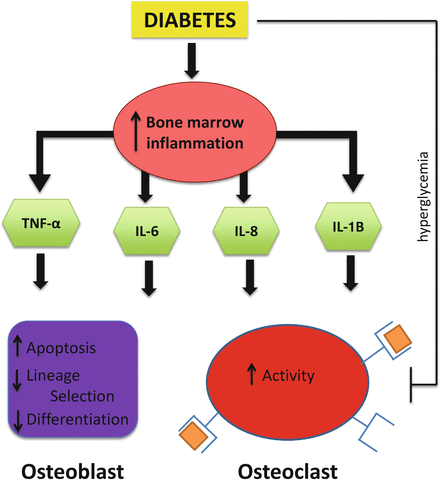
Fig. 8.3
Model for the role of inflammation in diabetes induced bone pathology. Both T1D and T2D conditions can increase bone marrow inflammation. The release of inflammatory cytokines into the bone marrow microenvironment by immune and bone cells can directly impact osteoblast and osteoclast activity which can potentially result in reduced bone turnover (T2D) or bone loss (T1D). TNF-α has extensively been shown to reduce osteoblast viability, lineage selection, and maturation. Similarly, IL-6, IL-8, and IL-1B can negatively affect osteoblast activity. Interestingly, while these cytokines are all capable of promoting osteoclast activity, conditions of diabetes such as hyperglycemia as well as expression of other inhibitory cytokines likely lead to the overall outcome of suppressed osteoclast activity typically seen in diabetes
Emerging evidence indicates that T1D directly compromises the function of the bone marrow [57]. Increased local cytokine levels in the bone marrow of mice under hyperglycemic conditions can disrupt hematopoiesis by shifting monocytes towards a pro-inflammatory phenotype [167, 172]. This change generated more monocytes and less endothelial progenitor cells (EPCs) in T1D, which contribute to the development of microvascular complications in this condition [166]. This finding was also evident in individuals with non-proliferative diabetic retinopathy [173]. In another experiment using Yorkshire pigs, after STZ-induction of diabetes a significant impairment of bone marrow and circulating EPCs as well as endothelial cells function were observed [174]. Chronic diabetes is also associated with dysfunction in the number and repopulation activity of HSPC, which is likely mediated by alterations in cytokines involved in stem cell renewal and maintenance [165]. Longstanding STZ-induced and spontaneous T1D mice further display impairment of colony-forming capability of the bone marrow and an increase in the number of Ly6Chi (pro-inflammatory) monocytes in the peripheral blood [166].
The bone marrow is also the source of cells that contribute to diabetes-associated inflammation. Bone marrow transplantation experiments done in both mice and rats, demonstrate that most of the extrapancreatic proinsulin producing cells (found in liver, adipose tissue, spleen, and thymus) originated from the bone marrow [168]. These cells were also demonstrated to produce the pro-inflammatory cytokine TNF-α which could contribute to damage of the target organs [168]. This suggests that the damage seen in organs such as bone during diabetes may be due to the irregularity in bone marrow stem cells.
Effect of T2D on Marrow Cells
T2D is not an autoimmune disease and is not always associated with bone loss [175]. However, there are changes in hematopoetic profiles. For example, T2D patients have 33 % less circulating progenitor cells (CPCs) and 40 % less circulating EPCs compared with healthy subjects [176]. Peripheral vascular complications of T2D mellitus are associated with an extensively low number of EPCs [176] which could result from alterations in bone marrow function. Altered Wnt signaling is also associated with T2D. A single nucleotide polymorphism locus in the Wnt5b gene conferred susceptibility to T2D by modifying adipocyte function [177]. Wnt signaling is also altered in immune cells. A link between cellular glucose sensing and the Wnt/b-catenin pathway was recently reported in two macrophage cell lines (J774.2 and RAW264.7 cells) [178]. T2D is also accompanied by an increased inflammatory state that can disrupt osteoclast function and survival [67]. However, a small study in T2D patients examining changes in bone turnover markers found that treatment with nonsteroidal anti-inflammatory drugs causes an increase in osteocalcin levels in men (not in women) and increases serum C-terminal telopeptide (s-CTx) [179]. This small study suggests that T2D inflammation decreases bone turnover.
Potential Mechanisms Contributing to Altered Bone Remodeling
Several mechanisms are thought to contribute to diabetes complications and the bone remodeling changes that occur with diabetes. Diabetes is associated with hyperglycemia, hyperlipidemia decreased insulin signaling, decreased IGF-1, oxidative stress, and inflammation; all of which can contribute to suppressed osteoblast activity among other additional regulators including amylin and incretins. We will discuss several of these mechanisms briefly below.
Hyperglycemia
Hyperglycemia has many negative effects on bone and bone formation. High glucose can result in the production of ROS [180, 181], induction of cellular osmotic responses [182, 183], and increased nonenzymatic glycosylation of proteins and DNA [144, 184–188] as well as suppress calciotropic hormones and growth factors [62, 144, 180–185, 187–189], all of which can lead to decreased osteoblast activity. In vitro studies exposing MC3T3-E1 cells (osteoblastic cell line) to high glucose conditions (30 mM) showed a significant decrease in markers of osteoblast maturation, such as alkaline phosphatase and osteocalcin [90, 190]. Even when MC3T3-E1 cells are exposed to hyperglycemic conditions for short time periods, 48 h, osteocalcin mRNA levels are significantly decreased compared to cells cultured under normal (5 mM) glucose levels [90]. MC3T3-E1 cells cultured in high glucose also show decreased mineralization [190, 191]. These in vitro results suggest hyperglycemia decreases osteoblast maturation.
Hyperglycemia also promotes MSC adipogenesis over osteogenesis [45, 60, 88–90]. Specifically, culturing primary rat osteoblasts under high glucose conditions (25.5 and 35.5 mM) increases levels of adipogenic markers (PPARy and aP2) and decreases levels of osteogenic markers (osteocalcin) [192]. A similar lineage switch was seen in human osteoblastic MG-63 cells cultured under high glucose conditions. The decrease in osteogenesis appears to be dependent on cAMP/protein kinase A (PKA)/extracellular signal-regulated kinase (ERK) signaling [193] and the PI3K/Akt signaling [192]. Hyperglycemia can also reduce osteoblast growth by 40 % [192] and induce osteoblast apoptosis. Both UMR and MC3T3-E1 osteoblasts undergo increased apoptosis when exposed for 24 h to AGE-BSA compared to unmodified BSA [194]. Thus, hyperglycemia can affect osteoblast activity through alterations in MSC lineage selection, osteoblast maturation, and osteoblast death.
Hyperglycemia decreases osteoclastogenesis in vitro. Using RAW264.7 cells and bone marrow macrophages, it was found that high levels of D(+)glucose inhibited osteoclast formation, ROS production (needed for bone resorption), caspase-3 activity (necessary for RANKL-induced differentiation) and migration to bone resorption pits [150]. In addition to this, osteoclasts derived from bone marrow cells extracted from mice which were exposed to high glucose had decreased mRNA expression of RANK and cathepsin K, decreased TRAP activity and decreased resorption activity [195]. Together, these studies indicate that a high glucose environment decreases osteoclast differentiation and activity.
Hyperglycemia also promotes a nonenzymatic reaction between glucose and proteins that results in AGEs. AGEs build up over time in diabetic tissues [196, 197] because they are irreversible modifications [198]. For example, T2D elderly patients display increased urine pentosidine (an AGE) which was associated with increased clinical fracture incidence and increased vertebral fracture prevalence in those with diabetes but not in those without diabetes [199]. Pentosidine, pyrraline and N(carboxymethyl)lysine (CML) are some of the more commonly characterized AGE products whose levels increase in tissues with collagen matrices, particularly pentosidine, under conditions of diabetes [200–202] and aging [199, 203, 204]. AGE modifications can change the structural and functional properties of proteins and correspondingly their levels correlate with diabetes complication severity, including glycosylated hemoglobin levels and bone loss [188, 201, 202, 205–209]. Type 1 collagen, the most abundant constituent of the bone matrix, is a target for AGE [210, 211], which can accumulate and alter the mechanical properties of bone, decreasing its toughness and could therefore contribute to skeletal fragility [212–214]. AGE increases collagen crosslinking, which can decrease bone strength and ductility, and increase brittleness [209, 214–218]. An increase in AGEs have been linked to a reduction in serum osteocalcin levels and are positively associated with levels of pentosidine which impairs the biomechanical properties of bone [216, 219–221]. AGEs can modify bone cell behavior by interacting with specific cellular receptors, such as RAGE (receptor for AGEs). When AGEs bind to RAGE on osteoclasts, NF-kB activity and ROS increase [222, 223]. RAGE is also expressed in osteoblasts and AGE treatment of osteoblasts causes decreased osteoblast maturation [184, 222]. In vitro and in vivo studies showed that AGEs increase calvarial periosteal cell apoptosis and induced apoptosis in primary cultures of human or neonatal rat osteoblastic cells. This effect was mediated through RAGE and increased p38 and c-Jun N-terminal kinase (JNK) [224]. The presence of AGE can also induce osteoblast apoptosis through activation of caspase-3 and caspase-8 [105, 108, 197]. Consistent with these findings, deficiency of RAGE results in an increased BMD compared with control mice [225]. Interestingly, in vitro studies using AGE-modified bone slices and osteoclastogenesis assays have shown that resorption was markedly inhibited. This was confirmed by a marked decrease in the release of type I collagen fragments generated by the collagenolytic enzymes secreted by osteoclasts [226]. This decrease in bone resorption is thought to be due not only to the alteration of the structural integrity of the bone matrix by AGEs, but also their effect on osteoclastic differentiation [226]. AGEs also increase inflammation by monocytes and macrophages due to upregulation of TNFα and IL-1β [227].
Hyperglycemia can also increase oxidative stress. Oxidative stress is triggered through two ways: the mitochondria becoming overloaded with glucose or through AGEs and Polyol signaling [88]. Oxidative DNA damage markers such as 8-hydroxydeoxyguanosine are increased in hyperglycemic diabetic mice but decreased when the mice are treated with insulin and become euglycemic [120, 228]. This oxidative stress marker is also elevated in T2D rats and could contribute to decreased osteoblast maturation based on the in vitro findings of oxidative stress negatively affecting osteoblast maturation [120, 122, 229, 230]. In addition, hyperglycemia-induced osteoblast apoptosis has been linked with an increase in oxidative stress [51, 231]. ER-stress activates the unfolded protein response mechanism leading to increased C/EBP homologous protein (CHOP) signaling and cellular apoptosis [51]. T1D rats showed an increase in CHOP positive osteoblasts in the femurs which also showed decreased bone mineral density [51].
Wnt Signaling
An important regulator of bone formation is Wnt10b [37, 232]. Wnt10b is critical for promoting MSCs toward the osteoblast lineage [232], enhancing osteoblast maturation and suppressing osteoblast apoptosis. Wnt10b activity increases expression of osteogenic transcription factors such as Runx2, Dlx5, and osterix [37]. In addition, targeted overexpression of Wnt10b in mouse bone leads to increased trabecular bone density [234], while Wnt10b knockout mice display significant bone loss [37]. Wnt10b binds its receptors LRP5, LRP6 and frizzled to increase intracellular beta-catenin levels and stimulate gene expression through TCF/LEF transcription factor binding to gene promoter regions. Alterations in the level or in the activity of LRP5 markedly affect bone density. Specifically, activating mutations lead to increased bone density [235] while inactivating mutations or LRP5 deficiency results in osteoporosis [35, 37, 236]. Similarly LRP5 mutations in humans can increase or decrease bone density [237].
Given that diabetes is marked by reduced bone remodeling and reduced osteoblast activity, a role for altered Wnt signaling has been suggested. It was recently found that TNFα transgenic mice display decreased Wnt activity implying a potential interaction between TNFα and the Wnt signaling pathway [238]. As noted previously TNF α is increased with diabetes [227, 228] and its expression is elevated in T1D mouse bone [72]. Correspondingly, Wnt10b expression is decreased in T1D bone (unpublished data, McCabe) suggesting that Wnt10b downregulation could contribute to the T1D bone phenotype [239, 240]. This is supported by decreased β-catenin staining of osteocytes and osteoblasts in diabetic mouse bones [241]. Interestingly, sclerostin, a circulating inhibitor of Wnt signaling made by osteocytes, is demonstrated to be elevated in T2D patient serum [242]. This could function to suppress Wnt10b and overall bone remodeling in T2D. Future studies will further determine the role of Wnts and other regulators of bone remodeling such as BMPs in the pathophysiology of diabetic bone.
Insulin and IGF-1 Signaling
Because this is already discussed in Chap. 1, we will only briefly discuss the role of insulin and insulin growth factor-1 (IGF-1) changes in diabetes that lead to altered osteoblast activity. Osteoblasts express insulin receptors and can readily respond to insulin treatment [33]. IGF-1 binds to insulin growth factor-1 receptors (IGF1R) and can activate osteoblast maturation, proliferation, and increased matrix generation [244]. Bone resorption is also regulated by IGF-1 acting through osteoblasts [244].
A hormone co-secreted with insulin, amylin is also suppressed in TD1 [48, 145]. IGF-1 has been reported to be decreased in T1D patients and animal models [245, 246]. Decreased IGF-1 in serum has been linked to the reduction in bone mineral density, increased adiposity in bone marrow, and decreased osteoblast differentiation. IGF-1 treatment in diabetic rats was able to increase the mineral apposition rate (rate at which new bone is formed) when compared to diabetic controls [65].
To determine if the lack of insulin is responsible for bone loss, insulin receptor knockout mice were analyzed for osteogenic and adipogenic markers. There was no change in osteogenic markers when compared to the control mice but adipogenic markers were decreased [62, 72, 79]. To analyze the effects of insulin-resistance (T2D) on osteoblasts, a mouse strain was generated to reproduce osteoblast insulin-resistance [247]. These mice were then fed a high-fat diet to induce T2D. Circulating osteocalcin levels were decreased and glucose intolerance worsened. The results implicate osteoblast insulin signaling in whole-body glucose homeostasis. Mice that lack insulin receptor signaling in osteoblasts exhibit either no change in osteoclast number [44] or a marked decrease in bone resorption markers [43]. The latter suggests a role for osteoblast insulin signals in enhancing resorption, consistent with studies demonstrating reduced or unaltered resorption in humans and mouse models of T1D. In both studies, OPG was increased and may be involved in suppressing osteoclast activity in T1 diabetics similar to other studies in T1-diabetic mice [67].
Role of Hyperlipidemia
Diabetes is associated with increased serum lipids. Hyperlipidemia has a number of adverse effects, including osteoporosis [250–254]. Hyperlipidemia results in increased amounts of LDL particles which can accumulate in the subendothelial space and are oxidatively modified to increase the generation of lipid oxidation products [255]. These lipid oxidation products can accumulate in bone [256]. Studies examining the effect of hyperlipidemia on bone health revealed increased levels of parathyroid hormone, TNF-α, calcium and phosphorus, and carboxyl-terminal collagen cross-links (a marker of bone resorption) [257]. Diabetic patients exhibit changes in serum lipid profiles along with hyperlipidemia linked with a decrease in metabolic control [258–260]. In female T1D patients, there was a significant correlation between increased serum lipid levels and decreased bone density [96]. Serum lipids can serve as ligands to activate PPARγ2 and promote altered MSC lineage selection and thus could play a role in suppressing osteoblast activity. However, in T1D mice, inhibition of PPARγ2 activity prevented hyperlipidemia but did not prevent bone loss [79]. This suggests that hyperlipidemia may not play a major role in T1D bone loss but could contribute to the altered lineage selection of MSC.
Studies examining the role of hyperlipidemia during osteoclastogenesis utilized pre-osteoclasts from hyperlipidemic mice (LDLR−/− mice) and control mice. After induction with M-CSF and RANKL, there was an increased number of resorption pits indicating an increase in osteoclastic activity [256]. In addition to this, there was also an increase in TRAP activity, indicating increased osteoclast differentiation [256]. Another study using LDLR−/− mice showed that serum levels of CTX, a marker for bone resorption was significantly increased in hyperlipidemic mice [257]. Together, these studies indicate a role for hyperlipidemia in osteoclast differentiation and activation. Although hyperlipidemia is associated with diabetes and is linked with osteoclast activity, hyperglycemia, the driving force of diabetes, decreases osteoclast activity and may work to override the effects of hyperlipidemia seen in patients.
Role of Calcitropic Hormones and Leptin
Studies are controversial regarding the effect of T1D on the serum level of vitamin D, which is key for enhancing intestinal calcium absorption and enhancing some stages of osteoblast maturation. While some studies report no changes in vitamin D levels, others report decreased levels in over 75 % of diabetic children and adults in the US northeast [261–263]. However, it is difficult to distinguish T1D associated vitamin D deficiency since more than 50 % of control subjects in winter months experience vitamin D deficiency [261]. In addition to the controversy about vitamin D deficiency, PTH levels in T1D patients are unchanged or decreased and in T2D patients are decreased [71, 72, 77, 264–266].
Another regulator of bone density is leptin [46]. Leptin is produced and secreted by adipocytes and has been shown to regulate lineage selection by stimulating MSC to differentiate into osteoblasts over adipocytes [189, 267, 268]. In T1D patients, serum leptin levels vary between patients [45, 269] although some studies report a decrease [270, 271]. Chronic treatment of hypoleptinemic T1D mice with leptin did not prevent bone loss but did reduce marrow adiposity [46]. T2D patients are even more complex with various factors affecting leptin levels including insulin secretion and resistance, gender and fat mass [33]. In vitro experiments show that leptin stimulates osteogenesis over adipogenesis [118, 189, 268].
Inflammation
Both obesity (which can result in T2D) and diabetic conditions can increase bone marrow inflammation. Inflammatory cytokines can directly impact osteoblast activity through changes in lineage selection, maturation, and death. T1D patients and mice have increased inflammation [108, 272–274], and elevated expression of inflammatory cytokines have been identified in T1D mouse bone [72]. Similarly, serum pro-inflammatory cytokines are increased in T2D patients [275–278]. Pro-apoptotic inflammatory cytokines like TNF-α induce osteoblast apoptosis and have been shown to be elevated in both T1D and T2D patients [46, 83, 147]. Elevated TNF-α mRNA levels are increased in T1D mouse whole crushed bone [83, 147], bone marrow [52], and gingival tissue [279]. When TNF-α binding to its receptor is inhibited by Pegsunercept, diabetes-associated osteoblast apoptosis is reduced [49, 280]. TNF-α and other pro-inflammatory cytokines such as IL-6 and IL-1 can also negatively affect osteoblast differentiation/activity and therefore suppress bone formation [281]. For example, in vitro studies show when cultured osteoblasts are treated with inflammatory cytokines (IFN-γ), there is a decrease in alkaline phosphatase activity [282].
Inflammation in the bone marrow favors expansion of osteoclasts and an increase in their activity. This increase in number can not only lead to bone loss, but also lead to impaired HSC maintenance and homeostasis [283]. Osteoclastic bone resorption can be activated by infection and inflammation. Because osteoclasts come from the same lineage as many immune cells, they express many innate immune cell receptors, such as toll-like receptors which can respond to antigen stimulation [284–286]. In addition to antigen stimulation, pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6 can induce osteoclast-mediated resorption [287, 288]. Studies in T1D mice have shown that induction of an immune response by the bacterial antigen LPS induces altered osteoclast differentiation with mature osteoclasts having increased bone resorption activity and increased release of osteoclastogenic factors (cathepsin K and MMP-9) compared to diabetic mice without LPS stimulation [289]. Cytokines secreted from adipose tissue, IL-6, IL-8, and TNFα, can both activate osteoclast bone resorption and decrease bone formation through the suppression of osteoblast differentiation [281]. While inflammatory conditions can directly activate osteoclasts, the effect of inflammation on osteoblasts can indirectly decreases osteoclast maturation and activity due to the extensive cross talk between the two cell types. A study examining the effects of bacterial-induced inflammation in T2D animals (ob/ob) showed that bone loss was not due to increased osteoclastogenesis but was dependent on the decreased levels of bone formation [112].
Summary
Diabetes affects bone remodeling through multiple mechanisms and alterations in multiple cell types. T1D and T2D have similar and different mechanisms at play that result ultimately in increased fracture risk. While we have mentioned several mechanisms here, there is still much to be learned and more contributors to the bone phenotype are likely.
Acknowledgments
NIH grants 5R01DK101050-02, 1RO1AT007695-01; SR—MSU Genetics Graduate Program; CV—MSU Biochemistry and Molecular Biology Graduate Program; NR—MSU Comparative Medicine and Integrative Biology Graduate Program.
References
1.
Hanley DA et al. Associations among disease conditions, bone mineral density, and prevalent vertebral deformities in men and women 50 years of age and older: cross-sectional results from the Canadian Multicentre Osteoporosis Study. J Bone Miner Res. 2003;18:784–90. doi:10.1359/jbmr.2003.18.4.784.PubMed
2.
Strotmeyer ES et al. Diabetes is associated independently of body composition with BMD and bone volume in older white and black men and women: the health, aging, and body composition study. J Bone Miner Res. 2004;19:1084–91. doi:10.1359/jbmr.040311.PubMed
3.
Bonds DE et al. Risk of fracture in women with type 2 diabetes: the Women’s Health Initiative Observational Study. J Clin Endocrinol Metab. 2006;91:3404–10. doi:10.1210/jc.2006-0614.PubMed
4.
Burghardt AJ et al. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:5045–55. doi:10.1210/jc.2010-0226.PubMedCentralPubMed
5.
Cummings SR et al. Risk-factors for hip fracture in white women. N Engl J Med. 1995;332:767–73. doi:10.1056/nejm199503233321202.PubMed
6.
Petit MA et al. Bone mass and strength in older men with type 2 diabetes: the Osteoporotic Fractures in Men Study. J Bone Miner Res. 2010;25:285–91. doi:10.1359/jbmr.090725.PubMedCentralPubMed
7.
Orlic D et al. Bone marrow stem cells regenerate infarcted myocardium. Pediatr Transplant. 2003;7 Suppl 3:86–8.PubMed
8.
Ianus A, Holz GG, Theise ND, Hussain MA. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest. 2003;111:843–50. doi:10.1172/Jci200316502.PubMedCentralPubMed
9.
Krause DS et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77.PubMed
10.
Semont A et al. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ. 2010;17:952–61. doi:10.1038/cdd.2009.187.PubMed
11.
Little RD et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70:11–9. doi:10.1086/338450.PubMedCentralPubMed
12.
Lian JB, Stein GS, Stein JL, van Wijnen AJ. Osteocalcin gene promoter: unlocking the secrets for regulation of osteoblast growth and differentiation. J Cell Biochem. 1998;30–31:62–72.
13.
Cook, David, and Paul Genever. Regulation of mesenchymal stem cell differentiation. Transcriptional and Translational Regulation of Stem Cells. Springer Netherlands, 2013;213–229.
14.
Nuttall ME, Gimble JM. Controlling the balance between osteoblastogenesis and adipogenesis and the consequent therapeutic implications. Curr Opin Pharmacol. 2004;4:290–4. doi:10.1016/j.coph.2004.03.002.PubMed
15.
Knouff C, Auwerx J. Peroxisome proliferator-activated receptor-gamma calls for activation in moderation: lessons from genetics and pharmacology. Endocr Rev. 2004;25:899–918. doi:10.1210/er.2003-0036.PubMed
16.
Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma 2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell. 2004;3:379–89. doi:10.1111/j.1474-9728.2004.00127.x.PubMedCentralPubMed
17.
Jilka RL, Weinstein RS, Takahashi K, Parfitt AM, Manolagas SC. Linkage of decreased bone mass with impaired osteoblastogenesis in a murine model of accelerated senescence. J Clin Invest. 1996;97:1732–40. doi:10.1172/jci118600.PubMedCentralPubMed
18.
Kajkenova O et al. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997;12:1772–9. doi:10.1359/jbmr.1997.12.11.1772.PubMed
19.
Verma S, Rajaratnam JH, Denton J, Hoyland JA, Byers RJ. Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J Clin Pathol. 2002;55:693–8. doi:10.1136/jcp.55.9.693.PubMedCentralPubMed
20.
Ferre P. The biology of peroxisome proliferator—activated receptors—relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53:S43–50. doi:10.2337/diabetes.53.2007.S43.PubMed
21.
Komori T. Regulation of osteoblast differentiation by transcription factors. J Cell Biochem. 2006;99:1233–9. doi:10.1002/jcb.20958.PubMed
22.
Lecka-Czernik B et al. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPAR gamma 2. J Cell Biochem. 1999;74:357–71. doi:10.1002/(sici)1097-4644(19990901)74:3<357::aid-jcb5>3.0.co;2-7.PubMed
23.
Picard F, Auwerx J. PPAR gamma and glucose homeostasis. Annu Rev Nutr. 2002;22:167–97. doi:10.1146/annurev.nutr.22.010402.102808.PubMed
24.
Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145:401–6.PubMedCentralPubMed
25.
Ali AA et al. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–35.PubMed
26.
Chuang CC, Yang RS, Tsai KS, Ho FM, Liu SH. Hyperglycemia enhances adipogenic induction of lipid accumulation: involvement of extracellular signal-regulated protein kinase 1/2, phosphoinositide 3-kinase/Akt, and peroxisome proliferator-activated receptor gamma signaling. Endocrinology. 2007;148:4267–75. doi:10.1210/en.2007-0179.PubMed
27.
Cho SW et al. Osteoblast-targeted overexpression of PPAR gamma inhibited bone mass gain in male mice and accelerated ovariectomy-induced bone loss in female mice. J Bone Miner Res. 2011;26:1939–52. doi:10.1002/jbmr.366.PubMed
28.
Akune T et al. PPAR gamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55. doi:10.1172/jci200419900.PubMedCentralPubMed
29.
Cock TA et al. Enhanced bone formation in lipodystrophic PPARgamma(hyp/hyp) mice relocates haematopoiesis to the spleen. EMBO Rep. 2004;5:1007–12. doi:10.1038/sj.embor.7400254.PubMedCentralPubMed
30.
Diascro DD et al. High fatty acid content in rabbit serum is responsible for the differentiation of osteoblasts into adipocyte-like cells. J Bone Miner Res. 1998;13:96–106. doi:10.1359/jbmr.1998.13.1.96.PubMed
31.
Lecka-Czernik B et al. Divergent effects of selective peroxisome proliferator-activated receptor-gamma 2 ligands on adipocyte versus osteoblast differentiation. Endocrinology. 2002;143:2376–84.PubMed
32.
Napoli N et al. The alliance of mesenchymal stem cells, bone, and diabetes. Int J Endocrinol. 2014;2014:690783. doi:10.1155/2014/690783.PubMedCentralPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


