34.1
Introduction
For decades the skeleton was viewed as a relatively isolated organ with little connection to other tissues in the body or to metabolic homeostasis. Certainly its role as a major reservoir for calcium emphasized its overall importance in essential cellular functions, but little was known about its role as an endocrine organ. There has been a convergence of evidence from both epidemiologic and basic studies suggesting that adipose and calcified tissue not only share the same progenitor cell but communicate and are integrated through the central nervous system (CNS) . As such, a new metabolic paradigm has emerged linking the skeleton with other organs as both an integrator of intercellular signals and a target for endocrine, paracrine, and autocrine factors. Secreted factors such as fibroblast growth factor-23 and sclerostin from osteocytes, and osteocalcin from osteoblasts provide important signals for phosphate balance, energy utilization, and insulin secretion and sensitivity. Moreover, a major role for insulin in skeletal remodeling has emerged providing further evidence that bone is both a target and source for metabolic regulation . In addition, epidemiologic studies have suggested that there is a complex relationship between total adipose tissue and bone mass . Earlier paradigms supported a positive role for adiposity in the prevention of fractures and protection against bone loss due in part to increased aromatization of testosterone and to enhanced gravitational forces on cortical bone . However, emerging data demonstrate that obesity may be a risk factor for osteoporosis in men, and that the metabolic syndrome in women is associated with reduced trabecular bone. In this chapter, we will focus on emerging lines of evidence demonstrating the dynamic nature of the skeleton relative to adipose tissue and the pathophysiologic implications of changes in bone or fat relative to two chronic diseases, obesity, and osteoporosis.
34.2
Bone and fat—epidemiological studies
Body composition is determined by the relative amount of lean and fat mass, the former primarily due to muscle and bone mass. Both compartments have a major genetic component and thus both osteoporosis and obesity are heritable and genetically correlated . Previous candidate gene and bivariate association studies have suggested some common genes for obesity and osteoporosis such as Runx2 . This is not surprising since mesenchymal stromal cells are the precursors for both osteoblasts and adipocytes. However, there may also be other factors that confound the association. For example, the amount of adipose tissue as a function of weight has a definite effect on the load placed on the skeleton. CNS signals such as sympathetic tone cause lipolysis and also enhance bone resorption while suppressing bone formation. Similarly, neuropeptide Y and its receptor Y1 promote lipogenesis both centrally and peripherally, while at the same time inhibiting bone formation .
The commonality between the origins of fat and skeletal tissue as well as their shared control has led to several large-scale observational (but not longitudinal) studies of the relationship between bone and fat. In England, for example, there was an extremely high prevalence of obesity among postmenopausal women with fragility fractures . In GLOW (Global Longitudinal Registry of Osteoporosis in Women), a large multinational study, obesity was not protective against fractures among postmenopausal women and in fact was associated with an increased risk of ankle and upper leg fractures ( Table 34.1 ) . Similarly Premaor et al. reported that in the Study of Osteoporotic Fractures, women who were obese and had fractures exhibited lower bone mineral density (BMD) than obese individuals without fractures, suggesting that fractures in obese individuals have the characteristics of fragility fractures in normal or lightweight individuals . In children, several studies have demonstrated that obesity is a risk factor for radial fractures particularly during times of rapid skeletal growth .
| Prevalent fracture at baseline [ n (%)] | Incident fracture in year 1 or 2 [ n (%)] | |
|---|---|---|
| Obese BMI >30 kg/m 2 | 3146 (23) | 657 (6.4) |
| Nonobese BMI 18.5–30 kg/m 2 | 10,272 (24) | 2278 (6.8) |
| Underweight BMI <18.5 kg/m 2 | 344 (32) | 56 (7.3) |
| Total | 13,762 (24) | 2991 (6.7) |
In respect to BMD a study in Korean men and women found that fat mass was inversely associated with bone mineral content after controlling for mechanical loading, although the location of excess adipose tissue may also contribute to confounding relative to bone mass . For example, Gilsanz et al. demonstrated that increased subcutaneous fat was associated with higher cortical bone indices and polar moment of inertia compared to other fat depots . In contrast, Bhupathiraju et al. showed that in Puerto Rican adults, increased abdominal obesity was associated with low bone mass at three femoral sites . In a cross-sectional study of patients with type 2 diabetes (T2D), insulin levels were inversely related to periosteal circumference and areal BMD and were related to intramuscular fat suggesting a lipotoxic effect on the skeleton . Bredella et al. demonstrated that in premenopausal women visceral adipose tissue measured by quantitative computer tomography (QCT) was inversely associated with volumetric BMD in the spine, while muscle area and density were positively associated with volumetric BMD .
In conclusion, the totality of evidence from epidemiologic studies would suggest that although adipose tissue may exert some protective effects on the skeleton due to the local generation of estradiol from testosterone and a gravitational stimulus to the periosteum, it may also cause deleterious changes in bone microarchitecture and strength, possibly through the elaboration of adipokines and cytokines acting in an endocrine manner. To define the relationships noted in epidemiologic studies between bone and fat, a clearer understanding of adipose tissue physiology and adipocyte origin is necessary.
34.3
Physiology of adipose tissue and energy expenditure
Adipocytic precursors originate from mesenchymal stem cells (MSCs) (i.e., the stromal vascular fraction) in the bone marrow and adipose depots around the body. The differentiation of these cells occurs in a well-defined but complex and tightly regulated process (see later) resulting in adult fat cells. Although there is a constant flux of substrate in and out of adipocytes, terminally differentiated cells store fatty acids and expand that storage during times of energy excess. This occurs principally through insulin stimulation and peroxisome proliferator–activated receptor gamma (PPARγ) activation by endogenous ligands. Adipocyte expansion represents the major process whereby fat tissue increases in size; in contrast, proliferation of preadipocytic cells from either the stromal vascular fraction or the bone marrow accounts for less than 10% of the increase in adipose tissue that occurs with obesity.
There are two types of fat depots in mammals: white and brown adipose tissue (WAT and BAT). The former is predominant in mammals and comprises between 95% and 99% of all adipose depots, whereas preformed BAT is present only in very localized interscapular depots. The structural and functional differences between these two tissue types are remarkable. BAT in contrast to WAT contains small multiloculated adipocytes that possess large numbers of mitochondria. WAT, in contrast, is composed of large single cells with lipid vacuoles surrounded by a simple membrane and few mitochondria. WAT stores triglycerides and fatty acids for use as an energy source to generate adenosine triphosphate (ATP), while BAT is purely a thermogenic organ responsible for the process of nonshivering thermogenesis, a means to maintain body temperature efficiently without muscle fatigue. Heat is generated in BAT through proton transfer out of the mitochondrial membrane, rather than by electron transport, the process that leads to hydrolysis of ATP. This shift from energy generation to heat production without utilizing muscle shivering is accomplished through upregulation of uncoupling protein 1 (UCP1), a gene expressed only by BAT and induced by sympathetic neurons that are activated with cold temperatures. BAT is found mostly in newborns and younger animals and diminishes with advancing age, although it is not clear whether those changes are related to apoptosis, atrophy, or metabolic dysfunction. A third type of adipocyte, the “bright cell,” is a white fat cell that becomes brown-like (i.e., thermogenic) in its function. These cells are also under the influence of sympathetic tone and express many of the markers of differentiated BAT. The importance of defining these two types of adipose tissue (BAT and WAT) underlies the complex relationship of fat mass to bone mass and skeletal remodeling. For example, increased intraabdominal fat has been negatively associated with volumetric bone volume fraction in mice and humans, whereas the volume of BAT measured by positron emission tomography scanning in humans, and weights in mice, has recently been positively associated with bone mass (Bredella, personal communication, 2012).
Cellular processes that require energy stimulate hydrolysis of triglycerides into fatty acids and glycerol. Humans and animals use energy to support diverse physiological and behavioral processes. Energy consumption in cells, such as protein synthesis and the maintenance of ion gradients, is supplied only by the conversion of ATP to adenosine diphosphate and inorganic phosphate . This is done in several ways. First, diverse substrates (proteins, fats, and carbohydrates) are processed into precursor molecules—the most important of which are pyruvate and acetyl-coenzyme A, common points in both glucose and fatty acid degradation pathways. Second, these substrates feed into the tricarboxylic acid cycle, which utilizes the energy in the molecules to convert NAD+ to NADH by trapping electrons. NADH carries the energy rich electron and donates it to a series of proteins embedded on the inner mitochondrial membrane called the electron transport chain (ETC). The ETC functions to capture the potential energy in electrons slowly and use it to pump protons from the mitochondrial matrix into the mitochondrial intermembrane space. These protons then pass back through the mitochondrial membrane via ATP synthase, and their potential energy is trapped to generate ATP. At the end of the ETC the electrons are combined with oxygen and protons to form “metabolic” water, which explains the need for oxygen to generate energy.
Even at complete rest, individuals still burn a substantial amount of energy. This energy expenditure is generally called the resting energy expenditure or the resting metabolic rate. It is controlled by signals from the brain via both central and peripheral factors. Outflow from the sympathetic nervous system regulates adipocytic function and is the final common pathway for metabolic balance. Basal energy expenditure or basal metabolic rate (BMR) is defined as the energy expenditure of an individual animal or human at rest (i.e., not physically active) in a supine posture, in a postabsorptive state (i.e., sufficiently long after feeding for the thermal effect of food to have disappeared), and at a thermoneutral temperature—that is, at or slightly above the lower critical temperature. BMR is known to vary between the active and inactive phases of the daily cycle, independent of the activity being performed .
Although it was long known that linear growth required substantial energy, it is now apparent that bone remodeling, the process of maintaining skeletal mass, also demands substrate availability. Both osteoclasts and osteoblasts utilize glucose and free fatty acids (FFAs) in order to accomplish their goals of resorbing and subsequently forming new bone. Hence, substrates from adipocytes are intimately tied to activities in the bone multicellular unit. The details of how this is accomplished have not been clearly delineated but mitochondrial function is essential for all cells in the bone-remodeling unit. In conditions such as age-related osteoporosis, or estrogen deprivation, the accumulation of reactive oxygen species hinders mitochondrial function by limiting maximal respiration; this could ultimately result in impaired bone turnover. Remodeling of adipose tissue also occurs and is accelerated with aging and following ovariectomy, almost certainly as a result of changes in the central control over metabolism. This results in increased intraabdominal and marrow fat mass at the expense of subcutaneous adipose depots.
In sum, energy needs for cellular homeostasis require ready sources of substrates, often provided by hydrolysis of stored triglycerides and glycogen through an intact mitochondrial system. Triglycerides are converted to FFAs, which represent both a fuel for all cells and a building block for intracellular fat storage in WAT. Intracellular triglycerides are also utilized by BAT and in mice, repetitive cold exposure leads to triglyceride depletion with subsequent utilization of circulating lipoproteins. In contrast, clearance of triglycerides is impaired in the states of obesity and insulin resistance. Thus there is a complex relationship between states of energy excess (i.e., obesity), energy insufficiency, and bone turnover, which can be traced to the release of adipokines and cytokines as well as FFAs.
34.4
Adipocytic and osteoblastic differentiation
Adipogenesis is a complex and integrated process in which several transcription factors work in a context-specific manner . The molecular and cellular mechanisms of adipocyte differentiation have been widely and extensively studied using in vitro and in vivo models. The initial step of adipogenesis is the lineage commitment of MSCs into preadipocytes. This occurs in the stromal vascular fraction of adipose depots, and in the bone marrow. Following the expansion of preadipocytes, MSCs differentiate into mature adipocytes under the tight control of multiple transcription factors including C/enhancer binding protein (EBP)β/Δ and PPARγ. Among these, PPARγ, a nuclear receptor and transcription factor, plays a central role in adipogenesis as evidenced by the finding that the loss of PPARγ in mouse embryonic fibroblasts leads to a complete absence of adipogenic capacity . In vitro, multiple transcription factors and coregulators have been implicated as modulators of the expression and function of PPARγ. For example, differentiation of 3T3-L1 cells, a well-recognized cell line that is used to recapitulate adipogenesis in vitro, is regulated by the integration of several transcription factors, including the C/EBP family, C/EBPβ, and Δ. These factors stimulate Ppar γ transcription by directly binding to the promoter region . Increased expression of Ppar γ activates the expression of another member of the C/EBP family, C/EBPα, which, in turn, enhances the expression of PPARγ. Partial loss of function of C/EBPα (i.e., a hypomorph) results in a mouse (A-Zip) with very little adipose tissue including virtually none in the bone marrow. Surprisingly, these mice also are insulin resistant. Thus the C/EBP family is critical for the induction of PPARγ in vitro. However, in vivo adipogenesis is much more complex and requires other transcriptional factors and cofactors for PPARγ regulation in part because PPARγ expression is maintained in adipose tissue of mice lacking C/EBPβ and/or delta .
34.5
The central role of peroxisome proliferator–activated receptor-gamma
PPARγ is a member of the PPAR family of transcriptional factors, which has multiple roles not only in cell fate determination but also in lipid biosynthesis, mitochondrial biogenesis, inflammation, neoplastic growth, and insulin sensitivity . PPARγ is composed of four variants including PPARγ1 and 2, the two major forms of PPARγ protein, which are produced by differential usage of promoters and alternative splicing. PPARγ1 is widely expressed in many tissues, including the liver, skeletal muscle, adipose tissue, and bone, while expression of PPARγ2, which possesses 30 additional amino acids in its N-terminus compared to PPARγ1, is restricted to adipogenic cells. Importantly, several lines of evidence demonstrate the existence of PPARγ2 in marrow stromal cells prior to their differentiation into adipocytes . PPARγ dimerizes with the retinoic X receptor alfa (RXRα) and activates transcription of target genes although a number of transcription factors and coactivators that are also involved in the regulation of Ppar γ expression and function. As described earlier, the C/EBP family, C/EBPα, β, and Δ, play a pivotal role in adipogenesis in cooperation with PPARγ. PPARγ expression and function are also regulated by other transcription factors during adipogenesis, including Srebp-1c, KLF5, KLF15, Zfp423, and early B cell factor, (Ebf1), while KLF-2 and GATA2/3 negatively regulates PPARγ expression .
Naturally occurring substances such as fatty acids and metabolites of arachidonic acids are potential in vivo candidates for PPARγ ligands . 15-deoxy-Δ 12,14 -prostaglandin J 2 (15d-PGJ2) is one such endogenous PPARγ ligand which is derived from arachidonic acid, although it still remains to be clarified whether 15d-PGJ2 is functional in terms of activating PPARγ in vivo . In addition to these, a synthetic class of compounds, the thiazolidinediones (TZD), are potent exogenous ligands for PPARγ and have been widely used in the treatment of diabetes .
PPARγ transcriptional activity is regulated by histone modification as well as ligand availability. In the absence of ligands, corepressors such as NCoR and SMRT as well as histone deacetylases (HDAC) are recruited to the protein complex of PPARγ, thereby forcing PPARγ to be transcriptionally silent . In contrast, when ligands are available, these proteins are dissociated from PPARγ protein complex machinery, and coactivators such as CBP and histone acetyltransferase (HAT) are recruited. Another level of regulation is created by the histone methylation by histone methyltransferase. Noncanonical Wnt pathways activated by Wnt5a have been shown to suppress PPARγ transcriptional activation through the histone methyltransferase, SETDB1 (SET domain bifurcated 1) .
Posttranscriptional modifications of PPARγ are also important components in regulating PPARγ activity. PPARγ2 has been shown to be phosphorylated at serine 112 by secretory factors, including epidermal growth factor and platelet-derived growth factor, resulting in the impairment of PPARγ2 transcriptional activity . In contrast, phosphorylation at serine 273 by Cdk5 enhances adipogenesis and has been linked to greater fat deposition and suppression of adiponectin . Agents that block Cdk5 phosphorylation including the TZD rosiglitazone have been demonstrated to enhance insulin sensitivity through this mechanism. Newer agents that block Cdk5 phosphorylation but do not have transcriptional activity have been described as pure insulin sensitizers . In addition to phosphorylation, sumoylation and ubiquitination confer a different level of posttranscriptional PPARγ regulation . In sum the importance of this nuclear receptor and transcriptional factor for cellular metabolic fate cannot be overstated both for adipocytes and for osteoblasts. Not surprisingly, there are multiple layers of regulatory control over PPARγ activity.
34.6
Peroxisome proliferator–activated receptor gamma and the skeleton
Osteoblasts and adipocytes share a common precursor (i.e., MSCs), which also serves as a source of progenitors for marrow fibroblasts, chondrocytes, and supporting stroma for hematopoietic cells Fig. 34.1 . Lineage allocation of marrow MSCs toward either adipocytes or osteoblasts is a finely tuned event in which lineage-specific transcription factors (such as Runx2 and osterix for osteoblasts and PPARγ2 for adipocytes) play critical roles. Importantly, in some but not all situations, lineage allocation of MSCs toward either of these cell types are considered to be mutually exclusive; that is, activation of PPARγ2 leads to enhanced adipogenesis at the expense of osteoblastogenesis and is associated with reduced expression and function of osteogenic transcription factors such as Dlx5, Msx2, Runx2, and Osterix . In line with this, suppression of PPARγ is reported to stimulate osteoblastogenesis and represses adipogenesis . These observations are also consistent with the findings from aged mice models where marrow adiposity is increased, bone mass is reduced, and these are associated with enhanced PPARγ2 expression . Similarly, haploinsufficiency or a hypomorphic mutation of PPARγ2 has been reported to show high bone mass and reduced marrow adiposity associated with increased osteoblast number and greater bone formation .
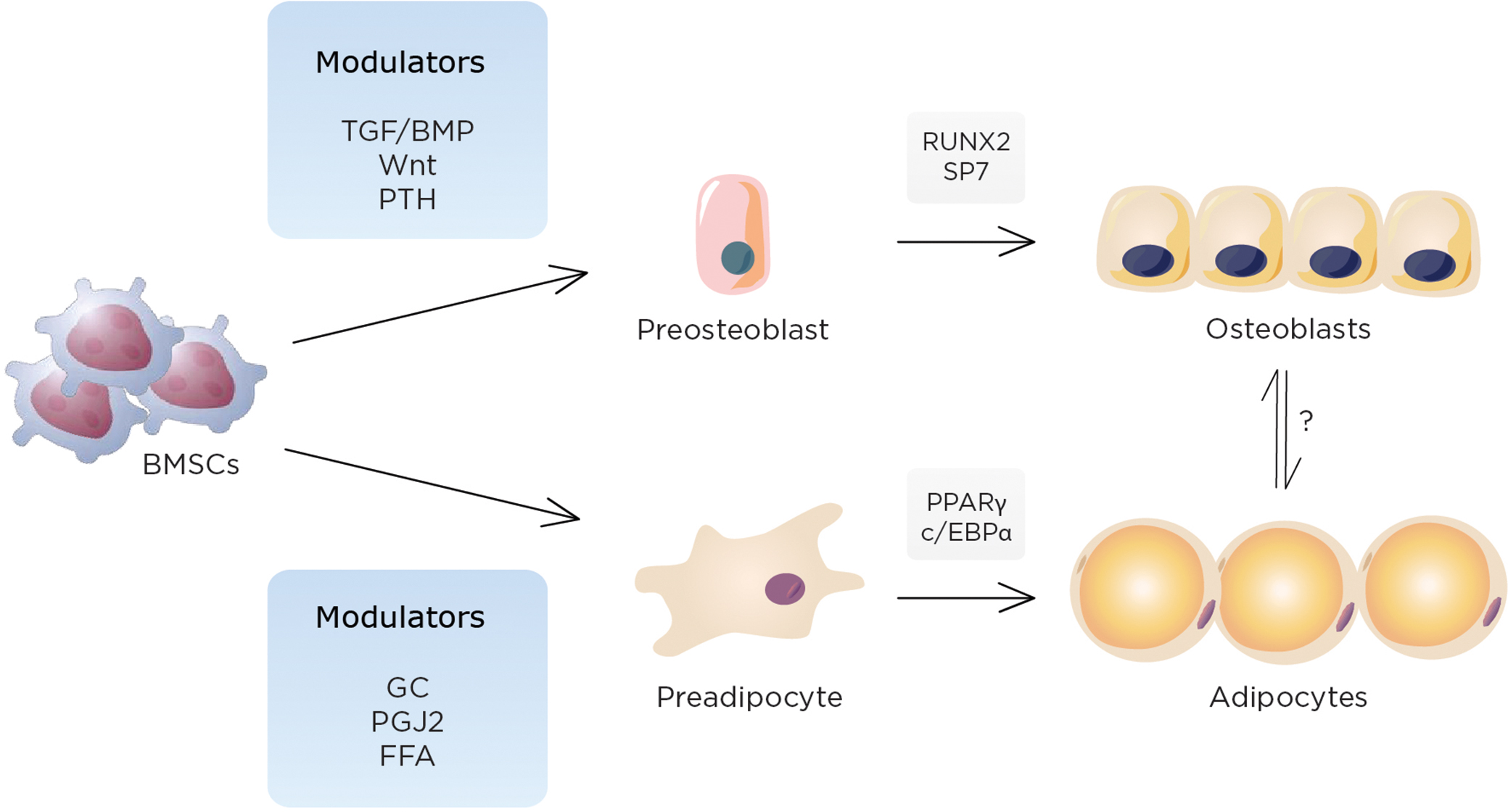
In addition to the pivotal role of PPARγ in lineage allocation of MSCs, mounting evidence indicates the involvement of PPARγ in osteoclast differentiation as well. For example, PPARγ activation has been shown to activate bone resorption in part through enhancing osteoclast differentiation by recruitment of another coactivator of PPARγ, PGC-1β . Furthermore, the effect of PPARγ activation on osteoclastogenesis could be in part mediated by the increased expression of Rankl in an age-dependent manner ; however, the exact role of PPARγ in osteoclastogenesis is still controversial and needs to be determined . In addition, we recently observed that conditional deletion of PPARγ in differentiated adipocytes using the aP2 promoter resulted in lipodystrophy by 26 weeks accompanied by high bone mass. Interestingly, these mice exhibit increased osteoclast activity suggesting that the phenotypic effects of PPARγ are highly context-specific with temporal effects from PPARγ deletion varying with the timing of inactivation.
The bone marrow is the source of MSCs necessary for both osteoblast and adipocyte differentiation. It is also the only site in the body where bone and fat cells reside adjacent to each other. Surprisingly, adipocytes have been reported in the bone marrow of mammals for many years, although few believed these cells were physiologically active. The developmental, mechanical, and physiological components of adipogenesis within the trabecular niche have become the focus of intense investigation. The function of marrow adipocytes has not been clearly delineated although it is likely that paracrine factors promote cell–cell interactions between osteoblasts and adipocytes, and this may impact bone remodeling. Aging, estrogen deprivation, glucocorticoids, and immobilization all increase marrow adiposity; whether this is at the expense of bone formation or a compensation for impaired osteoblast function remains to be determined.
34.7
Hormonal factors that regulate bone and energy metabolism
34.7.1
Intermediary metabolism and bone
Previously, energy metabolism was analyzed within the context of classical insulin-dependent tissues . In this respect the liver, the central component of glucose homeostasis, plays a preponderant role in glucose production during fasting, providing a constant fuel supply to the CNS . Muscle contributions were perceived by means of variation in the use of substrates, mainly FFA, in postabsorptive conditions, to have a pivotal position in glucose disposal after carbohydrate intake . From this perspective, adipose tissue was presumed to be a passive site for fat storage after feeding and a source of FFA and glycerol during fasting . Hormonal regulation was considered to be a simpler arrangement, placing insulin and the counter-regulatory hormones (glucagon, epinephrine, cortisol, and growth hormone) in diametrically opposed positions . In addition, it was recognized that several hormonal disorders (hyperthyroidism, hyperprolactinemia, hyperandrogenism, and primary hyperparathyroidism) were associated with insulin resistance, with only exercise and weight loss being able to increase insulin sensitivity . Curiously, all of these diseases also have profound impacts on the skeleton .
Great advances in clinical investigation have been made using the forearm model to measure peripheral glucose uptake and hepatic vein catheterization to determine glucose production and hyperinsulinemic clamp to evaluate insulin resistance . These investigations quantitatively revealed glucose disposal in muscle and liver in physiological and insulin-resistant states but were insufficient to uncover the cause of glucose intolerance. Moreover, metabolically active tissues such as intestine and bone were not considered in this estimate. Subsequently, landmark studies by Kahn et al. contributed to improve our knowledge about the molecular mechanisms of insulin resistance . In addition, these authors, using the Cre-lox system to disrupt the insulin receptor (IR) in mouse skeletal muscle, investigated the metabolic impact of severe muscle insulin resistance . Unexpectedly, these mice exhibited elevated fat mass, serum triglycerides, and FFA, but not high blood serum insulin or abnormal glucose tolerance. Thus the authors concluded that insulin resistance in muscle contributes to the altered fat metabolism associated with T2D, but tissues other than muscle appear to be more involved in insulin-regulated glucose disposal than previously recognized . This tenet has been initially confirmed by different studies showing that the functional scope of adipose tissue is not restricted to energy storage but is an active player in the emergence of insulin resistance . More recently, strong evidence has indicated that bone contains other elements that could modulate energy metabolism .
Adipose tissue plays a critical role in energy balance by synthesizing an array of factors that regulate satiety (leptin), insulin sensitivity (adiponectin and resistin), hormones (estrone), and proinflammatory molecules . As stated above, many or, more appropriately, all of these factors can directly or indirectly influence bone remodeling. The knowledge of the endocrine role of adipose tissue in normal physiology as well as pathophysiologic states is still superficial, especially because the peculiar switch in its secretion profile, as the adipocyte increases the storage of fat droplets, determines an adverse metabolic milieu. Obesity, the disorder of positive energy balance, is a common link between arterial hypertension, dyslipidemia, T2D, and ultimately cardiovascular disease. Therefore the obesity profile is a critical factor for the development of cardiometabolic risk factors associated with the metabolic syndrome . Despite being a risk factor for cardiovascular disease and diabetes mellitus, obesity has been thought to protect against osteoporosis. Body weight is positively associated with bone mass, the measurable component of bone strength . However, the insulating effect of adipose tissue on bone has been questioned, particularly in studies showing increased fracture incidence in obese old women as well as in adolescents . In addition, studies have demonstrated a differential impact of specific fat compartments on BMD with visceral adipose tissue having potential detrimental effects on BMD . It is apparent that the relationship of fat mass to bone mass is highly dependent on the sites being measured as well as the function of adipose depots.
The input of bone in the modulation of intermediary metabolism has been recognized in studies by Karsenty et al. and Clemens et al. . Osteocalcin, which is synthesized in osteoblasts, may be a link between hard tissue and the pancreas. Actually, nonmineralized and mineralized mesenchymal tissues are elements of integrative physiology by modulating and at the same time being influenced by energy metabolism. Not unexpectedly, insulin and vitamin D are hormonal agents for the integration between bone and energy metabolism . Their participation in this task will be specifically discussed later in this chapter. A failure in the proper action of these hormones results, to a variable extent, in the development of two highly prevalent and commonly overlapping diseases—diabetes mellitus and osteoporosis.
34.7.2
Insulin
Despite intensive studies the knowledge of the effects of insulin remains multifaceted and incomplete. Insulin orchestrates adipose tissue expansion and retraction as it adapts to a different nutrient supply. Alterations in adipose tissue remodeling provoked by overfeeding include infiltration of macrophages triggering an inflammatory status. Accordingly, large clusters of macrophage-related inflammatory genes have been reported to be significantly altered in hypertrophied adipose tissue . Macrophage accumulation in WAT is dependent on anatomical localization. There is twice as much infiltration in visceral as in subcutaneous WAT The proinflammatory molecules originating in visceral fat have been consistently associated with cardiovascular and hepatic diseases and more recently with decreased bone mass in adolescents . However, the impact of insulin resistance on bone remodeling and especially on the fate of bone marrow MSC is still to be determined.
In insulinopenic states such as a pharmacological model of type 1 diabetes mellitus, decreased bone volume was observed in trabecular bone of the tibia and femur. The decline in bone mass was responsive to parathyroid hormone (PTH) treatment. Osteocalcin and TRAP5 expression in bone of diabetic mice was lower and similar, respectively, compared to control mice, indicating the primordial impairment of bone formation in insulin deficiency . Further information was obtained in a well-designed study by Fulzele et al. . By silencing IRs specifically in osteoblasts, these authors observed that concomitant with decreased expression of IR, the primary osteoblasts exhibited enhanced expression of Twist2 and decreased expression of Runx2, a master differentiation factor for osteogenesis . Fulzele et al. concluded that IR signaling in osteoblasts controls osteoblast development and osteocalcin expression by suppressing the Runx2 inhibitor Twist2. Mice lacking IR in osteoblasts have low circulating undercarboxylated osteocalcin and reduced bone acquisition due to decreased bone formation and deficient numbers of osteoblasts. Moreover, these authors observed that, with age, these mice develop marked peripheral adiposity and hyperglycemia accompanied by severe glucose intolerance and insulin resistance . The metabolic abnormalities of these mice are improved by the infusion of exogenous undercarboxylated osteocalcin. In the same line of investigation, data obtained by Ferron et al. not only supported this hypothesis but also added new ingredients to the participation of bone in the control of glucose disposal. These authors suggested that the acidic bone microenvironment created by osteoclasts favors the decarboxylation of osteocalcin and potentiates the capacity of this molecule to stimulate β-cells to synthesize insulin . Therefore taken together, these results indicate the existence of a bone–pancreas endocrine loop through which insulin signaling in the osteoblasts ensures osteoblast differentiation and stimulates osteocalcin production, which in turn regulates insulin sensitivity and pancreatic insulin secretion to control glucose homeostasis.
In the bench-to-bed transition, type 1 diabetes mellitus corresponds to the clinical condition that might be used to validate the potential role of insulin in bone mass development and maintenance. Several studies have shown that type 1 diabetes mellitus is associated with osteopenia . In contrast the understanding of the influence of variations in insulin sensitivity on bone has been much more challenging. It is well known that exercise has a positive impact on bone mass and insulin sensitivity. Nevertheless, previous studies have shown that female runners exhibit lower lumbar spine BMD than controls . The frequency of oligo- and amenorrhea in the runner group was 30%. In addition, eumenorrheic swimmers had lower bone mass in femoral neck and trochanter not only compared to gymnasts but also compared to control females . These data indicate that the amelioration of insulin sensitivity is not sufficient to overcome the detrimental effect of estrogen deprivation and the long permanence in a weight-supported environment for bone mass.
Notwithstanding these data, the ultimate effect of insulin resistance on bone still remains to be determined. The first aspect needing clarification is how bone cells and their progenitors behave in conditions of insulin resistance. In most situations of reduced insulin sensitivity, compensatory hyperinsulinemia is a common outcome. Since insulin resistance is tissue- and pathway-specific, it is possible that, parallel to the reduced action of insulin in metabolic pathways, hyperinsulinemia may act by overstimulating other insulin targets in different cells and tissues. In addition, cross-talk between insulin and other hormones, such as luteinizing hormone (LH), angiotensin II (Ang II), epinephrine, growth hormone, and leptin, can modulate the final outcomes of insulin action . These events lead the organism to develop abnormal tissue responses and ultimately to the emergence of diseases such as hyperandrogenism and hypertension due to an inappropriate interrelationship between hyperinsulinemia and LH and Ang II, respectively.
Aging and glucocorticoid therapy, respectively, the natural and the drug-induced states of insulin resistance, have in common the tendency to weight gain and bone loss in association with increased content of bone marrow fat . Bredella et al. have shown by H 1 magnetic resonance imaging (MRI) spectroscopy that visceral fat correlates positively with bone marrow fat and negatively with volumetric BMD . Conversely, in anorexia nervosa, a primarily psychiatric disorder characterized by severe disturbance of eating behavior, there is normal insulin sensitivity accompanied by high serum levels of adiponectin . Despite the scarcity of overall body fat, these patients have higher bone marrow fat, as estimated by MRI spectroscopy, and decreased bone mass. The same pattern of bone composition is observed in mice submitted to caloric restriction . Finally, the insulin-sensitizing TZD drugs stimulate adipogenesis in the subcutaneous and bone marrow niche . In parallel to the improvement of glucose metabolism, on a long-term basis, individuals taking rosiglitazone show rapid bone loss and increased skeleton fragility . Taken together, these data reveal that there is not a linear relationship between bone marrow fat content and insulin sensitivity. The diverse pathways in the commitment of bone marrow MSC to osteoblasts or adipocytes most likely conceal key responses for the understanding of the etiopathogenesis of osteoporosis as well as T2D.
34.7.3
Vitamin D
At the turn of the last century, vitamin D deficiency was a well-characterized severe skeletal disease mainly affecting children. The clinical features, stereotypically designed by the presence of bowed legs and/or an undernourished state, were so conspicuous that diagnosis could be established based on clinical grounds. This outmoded picture contrasts with the current awareness that modest vitamin D deficiency may occur, affecting adults and the elderly in particular. Major degenerative disorders such as osteoporosis, T2D, arterial hypertension, and cancer may have been linked to vitamin D insufficiency in association studies, but as of 2020, there are no randomized controlled trials demonstrating that vitamin D supplementation can reduce chronic disease incidence .
The ubiquitous presence of the vitamin D receptor (VDR) as well as the cellular machinery for paracrine/autocrine synthesis of 1,25(OH) 2 D favor the multifunctional role of vitamin D. However, the synthesis process and action of vitamin D are so sophisticated and complex that we still have indirect and partial tools for the evaluation of vitamin D status . Lessons have been learned by using genetically engineered mice, especially regarding bone . For example, Panda et al. published insightful data using 1α-hydroxylase knockout mice, VDR knockout mice, and a combination of the two knockouts . These groups of mice were treated with either a high-calcium diet or a rescue diet with very high calcium and lactose or with 1,25(OH) 2 D. In 1α-hydroxylase knockout mice, physiological doses of 1,25(OH) 2 D corrected longitudinal bone growth to normal size. The high-calcium diet and the rescue diet could not correct longitudinal bone growth in the 1α-hydroxylase knockout animals. The longitudinal bone growth in the VDR knockouts could not be corrected by diets or 1,25(OH) 2 D. However, the rickets could be completely cured with the rescue diet containing a high calcium content and lactose . Thus it appears that for the healing of rickets neither 1α-hydroxylase nor VDR is necessary. Also these data indicate that dietary manipulation induce different phenotypic and that the mineralization of bone reflects ambient calcium and phosphorus levels rather than the direct actions of the 1,25(OH) 2 D/VDR system.
There are scarce data about the impact of vitamin D and its active form 1,25(OH) 2 D on the fate of bone marrow MSCs, but adipose tissue seems to be a singular site for the study and understanding of the endocrine, paracrine/autocrine effect of this secosteroid. Before hepatic and renal hydroxylation, vitamin D shows an increased lipophilic property; as a consequence, obesity-associated low serum 25-(OH)D is credited to adipose tissue sequestration . The status of vitamin D “deprivation” in obesity is further supported by the frequent description of high serum levels of PTH. Not unexpectedly, secondary hyperparathyroidism enhances the synthesis of 1,25(OH) 2 D . Results showing that 1α-hydroxylase and VDR knockout mice exhibit a lean phenotype and greater insulin sensitivity have raised several questions about the relationship between the vitamin D complex and diseases: (1) do the increased levels of 1,25(OH) 2 D due to secondary hyperparathyroidism contribute to the maintenance of weight gain and insulin resistance in obesity? (2) as systemic 1,25(OH) 2 D levels increase in both conditions (vitamin D insufficiency and with an increasing supply of vitamin D), are the variations in serum 1,25(OH) 2 D levels responsible for the U-shape curve of high serum 25-(OH)D levels in respect to all-cause mortality .
There are concurrent data suggesting that impairment of the effect of 1,25(OH) 2 D is associated with a lean phenotype. In 2011 it was observed in VDR heterozygous mice that the influence of VDR on bone and body composition is both age- and gender-dependent . While female mice with this genotype show decreased bone mass and normal body composition, males exhibit lower adiposity and similar bone mass compared to their wild-type counterparts. Intriguingly, the lean phenotype seems to be an ongoing process starting after 8 weeks of age. In a new approach the VDR was further evaluated by using the aP2 gene promoter to target the expression of the human (h) VDR in adipocytes in mice . In contrast to VDR-null mice the aP2-h VDRTg (transgenic mouse model that specifically expresses human VDR gene in adipocytes) mice developed obesity without variation in food intake. The increase in fat mass was mainly due to markedly reduced energy expenditure, which was correlated with decreased mobility and reduced fatty acid β-oxidation and lipolysis in the adipose tissue of transgenic mice . Consistently, the expression of genes involved in the regulation of fatty acid transport, thermogenesis, and lipolysis were suppressed in the transgenic mice. These new results reinforce the role of the VDR in fat mass gain.
Indirect evidence of the influence of 1,25(OH) 2 D on human body weight has been suggested by the observation that a hypercalcemic diet favors weight loss . It was hypothesized that increased calcium intake inhibits PTH synthesis which impairs 1,25(OH) 2 D synthesis. At the cellular level, 1,25(OH) 2 D plays a direct role in the modulation of adipocyte intracellular ionized calcium ([Ca 2+ ]i) through its membrane receptor . Thereafter, [Ca 2+ ]i signaling mediates both increased lipogenesis and decreased lipolysis. The increase of [Ca 2+ ]i within murine and human adipocytes stimulates the expression and activity of fatty acid synthase, a step-limiting enzyme in lipogenesis . In addition to stimulating triacylglycerol synthesis, increased intracellular calcium inhibits lipolytic activity, favoring fat storage. The antilipolytic effect of [Ca 2+ ]i is attributed to a direct activation of phosphodiesterase 3B, resulting in a decrease in cyclic adenosine monophosphate (cAMP), reducing the ability of agonists to stimulate the phosphorylation and activation of hormone-sensitive lipase . Therefore a hypercalcemic diet-induced suppression of 1,25(OH) 2 D synthesis reduces lipogenesis activity and favors lipolysis in adipose tissue through its nonclassical receptor. In addition, 1,25(OH) 2 D may regulate uncoupling protein 2 (UCP2) expression in WAT through the genomic pathway . In vitro, human adipocytes treated with 1,25(OH) 2 D exhibit a decreased expression of UCP2 . Moreover, VDR disruption prevents the inhibitory effect of 1,25(OH) 2 D on adipocyte UCP2 expression. Taken together, these data suggest that there are redundant mechanisms for the stimulation of fat-accumulation induced by 1,25(OH) 2 D.
Paradoxically, human obesity is traditionally considered to be a condition associated with vitamin D deficiency. At present, there is no answer regarding the influence of vitamin D therapy on body weight gain in the obese population. Based on the above evidence, it can be suggested that serum 1,25(OH) 2 D levels should be considered in addition to the measurement of 25-(OH)D to determine ideal levels of vitamin D replacement.
34.8
Drug-induced changes in bone and fat
There are common genetic determinants for bone and fat, as well as a shared mesenchymal progenitor . Hence, drugs that affect either tissue could have an “off target” effect on the other. Moreover, both bone and fat are regulated by central neural mechanisms located in the brain stem and hypothalamus. Since antidepressants and antipsychotic drugs affect the CNS and are widely used across many age groups, it should not be surprising that other tissues could be affected adversely. This section will focus on the emerging evidence that second-generation antipsychotics (SGAs) and selective serotonin reuptake inhibitor (SSRI) can adversely affect bone and fat. Although glucocorticoids are the quintessential drugs that adversely affect bone, fat, and brain, their effects on bone are covered in a separate chapter (see Chapter 49: Human immunodeficiency virus and osteoporosis).
Osteoblasts and osteoclasts express 5-hydroxytryptamine (5-HT; serotonin) receptors and can be exposed to serotonin, an important neurotransmitter, through endocrine, paracrine, and autocrine pathways . Moreover, evidence that leptin, an adipocyte-originated molecule, interacts with neurons and neuropeptides [dopamine, serotonin, epinephrine (adrenaline), and histamine] highlights the regulatory role for the CNS on adipose tissue and the skeleton and their interrelationship .
In the 2000s, some psychiatric diseases, such as schizophrenia, have been associated with weight gain, bone loss, and fractures independent of drug treatment, reinforcing this link of bone, fat, and brain .
Drugs employed for psychiatric diseases may vary in their mechanisms of action, namely, through dopamine (D)2, histamine (H)1, 5-HT2 receptor antagonism, blockade of serotonin reuptake transporter (5-HTT), and adrenergic antagonism. Many of them, mainly atypical antipsychotics and SSRIs have been associated with adverse metabolic and osteogenic effects .
34.9
Second-generation antipsychotics
SGAs, often called “atypicals,” emerged in the 1990s and represented a great therapeutic advance in managing psychotic symptomatology in part because of the absence of large-scale extrapyramidal manifestations as well as greater effectiveness than first-generation typical antipsychotics, in the relief of negative, cognitive, and affective symptoms. Some of these drugs have been widely used for other clinical situations such as dementia, aggressive behavior and in children and adolescents for mood and behavior disorders and irritability associated with autism .
SGA use has been associated with excessive weight gain, insulin resistance, dyslipidemia, and possibly bone loss . Obesity and related disorders present a high risk of cardiovascular diseases, one of the most frequent causes of death in the world population. SGAs are mostly employed in three groups that are more vulnerable to bone loss: elderly people, schizophrenics, and growing children, and adolescents.
The mechanisms of metabolic changes are not well understood. These undesirable side effects cannot be totally separated from the effects of the underlying psychiatric disease. At the skeletal level, there is some evidence that SGAs are associated with low BMD and risk of fractures in adults, children, and adolescents . The same effects have been shown for rodents treated either orally or parenterally, although no significant body weight gain has been noted . Possible mechanisms for bone loss with chronic use of SGAs include dopaminergic blockade–induced hyperprolactinemia and/or central serotoninergic antagonism .
Hyperprolactinemia is one of the causes of secondary hypogonadism and may be present in some patients during SGA treatment. However, there is no uniform association either from SGAs with hyperprolactinemia nor from low BMD during SGAs treatment with hyperprolactinemia. In part confounding by hypoestrogenemia makes it difficult to define whether changes in prolactin are directly related to bone loss with treatment. Interestingly, hyperprolactinemia from a number of different causes has also been associated with weight gain and obesity. Rodent models of SGA treatment demonstrate bone loss but without a concomitant increase in body fat or weight, suggesting that there must be other underlying mechanisms to explain bone loss during this treatment .
Serotonin has a dual effect on bone metabolism. Centrally, serotonin may increase BMD due to changes in leptin, while peripherally it exerts a negative impact on bone mass, possibly through inhibition of Lrp5 . Some genetic experiments have suggested that CNS serotoninergic signaling prevails over that of peripheral serotonin in the influence on skeletal function. Therefore it has been hypothesized that central serotoninergic blockade could drive bone loss with atypical antipsychotic drugs use .
In young mice treated with risperidone a commonly prescribed SGA, there was significant trabecular bone loss. Hyperprolactinemia was not detected in the treated group and the negative skeletal consequences were associated with enhanced osteoclastic activity in vivo and in vitro. As in earlier studies, there was no change in weight among mice treated with risperidone relative to the control group. Interestingly, marrow adiposity was enhanced in the SGA treated group . Data for an increased risk of fractures in adolescent and young adults treated with risperidone or other SGAs are lacking, although one paper suggests that bone loss can occur in younger psychiatric subjects treated with SGAs .
34.10
Selective serotonin reuptake inhibitors
SSRIs are the first-line antidepressants used worldwide. Depression is a very frequent psychiatric disorder and has previously been associated with osteoporosis, possibly mediated by brain-to-bone sympathetic signaling . Moreover, weight gain is a frequently observed side effect of all the SSRIs. However, the relationship between lean and fat mass among SSRI users is complex.
Changes in CNS serotonin levels are proposed as one of the mechanisms by which SSRIs not only alleviate depression but also affect sympathetic tone. SSRIs prevent the reuptake of serotonin through the inhibition of the 5-HTT on the presynaptic neuron (central action) and on bone cells (peripheral action). At peripheral sites, serotonin restrains osteoblastic activity, leading to bone loss. This action may outweigh the skeletal benefits resulting from its enhanced central serotonin .
Several observational and some animal studies have found that the use of SSRIs has been associated with either bone loss or fracture risk. This association persists even after adjusting for other risk factors . However, there are no randomized trials to define absolute risk or to determine potential mechanisms.
In summary, bone and adipose tissue are interrelated in the maintenance of energy balance and mineral/bone remodeling. Adipose tissue has the primordial role of energy storage, but it also displays important endocrine roles, which are essential in the modulation of appetite and for the gonadal axis. The heterogeneity of adipose tissue plasticity is site dependent. It is well established that visceral fat accumulation is the part of the noxious process that ultimately leads to insulin resistance through adipokines mediation. Bone marrow fat accumulation also is a common occurrence in several conditions associated with osteoporosis, but the association of insulin resistance with osteoporosis still is to be delineated. Bone remodeling potentially contributes to glucose homeostasis. Osteocalcin can stimulate insulin secretion and in turn insulin signaling in osteoblasts leads to osteocalcin synthesis. The CNS takes part in this process, being responsive to leptin and thereby activating the sympathetic nervous system. Bone formation is negatively affected by β-adrenergic input. Serotonin may be an intermediary between leptin and the CNS. Primary psychiatric disorders and their treatment with SSRIs or SGA represent a clinical model whereby changes in adipose function can be related through the CNS to patterns of bone loss.
References
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


