AML, Acute myeloid leukemia.
∗ Determined among 1311 patients with de novo AML enrolled onto Study 8461 of the Cancer and Leukemia Group B.
† Partial tandem duplication of the MLL1.
‡ Three or more chromosomal aberrations in the absence of t(8;21), inv(16)/t(16;16), t(15;17), or t(9;11).
Source: Adapted from Grimwade D, Mrozek K. Hematology/Oncology. Clinics of North America. 2011, 25: 1135-1161.
Molecular Pathogenesis of AML
The most frequently mutated genes in AML encode transcription factors, which are typically implicated in chromosomal translocations that result in their inappropriate activation in the hematopoietic compartment, resulting in impairment of maturation and differentiation of myeloid cells (e.g., CBF translocations and RARα and MLL gene rearrangements) (Figure 28-1 ). Frequent types of mutations are those that confer a proliferative and survival advantage to cells (i.e., activating mutations). An example of the latter include mutations in KIT or FLT3 (Table 28-2 ). Mutations in AML are a concept in flux, both during the natural course of the disease and during the course of therapy (i.e., clonal evolution), as shown by whole-genome sequencing analyses performed in paired samples obtained at diagnosis and at relapse. Of note, in some cases, new mutations were found on relapse in the dominant clone in the primary leukemia sample, whereas in others, these newly acquired mutations occurred in small subclones of the founding clone, which resulted in expansion and clonal dominance in the relapse sample. Importantly, varying proportions of clonal cells from the founding clone persisted after chemotherapy in all cases.
Mutations Disrupting the Function of Transcription Factors in AML
Two different types of transcription factors play a major role in hematopoiesis and therefore in the pathogenesis of AML. The first group consists of master regulatory transcription factors, which, like AML1, are implicated in the development of all hematopoietic lineages. Impairment of the signaling stemming from these types of transcription factors results in complete hematopoietic failure. A second category of transcription factors is involved in the development of specific hematopoietic lineages. For instance, GATA-1 skews the development of hematopoietic progenitors toward the erythroid lineage, whereas C/EBPα promotes granulocytic differentiation.
PML-RARα Rearrangements
Translocations involving the retinoic acid receptor (RAR) locus on chromosome 17, such as t(15;17)(q22;q11), drive the pathogenesis of acute promyelocytic leukemia (APL), which comprises 10%-15% of all cases of adult AML. Such translocations produce the PML-RARα transcript that encodes a fusion protein containing most of the functional domains of RARα (including the RAR binding domain and the DNA binding domain) and the majority of the PML gene. The breakpoints within the PML gene cluster locate to three different regions referred to as breakpoint cluster region (bcr) 1, 2, and 3. In addition to t(15;17)(q22;q11), there are at least two other variant translocations involving RARα associated with the APL phenotype. These include t(11;17)(q23;q21) and t(5;17)(q35;q21), which lead to the fusion of the RAR? gene to the promyelocytic leukemia zinc finger (PLZF) and NPM1 genes, respectively. Transgenic mice expressing PML-RARα, NPM/RARα, or PLZF/RARα under the control of a human Cathepsin G exhibit an APL phenotype after a variably long latency. Comparison of gene expression profiles set by PML-RARα and PLZF-RARα have demonstrated the inhibition of genes involved in DNA repair, repression of myeloid transcriptional regulators, and activation of the WNT/Catenin and Jagged/NOTCH pathways, which promote self-renewal of leukemic cells. Patients with APL harboring PLZF-RARα fail to respond to all-trans retinoic acid (ATRA), although, paradoxically, both PML-RARα and PLZF-RARα contain identical RAR sequences and inhibit ATRA-induced gene transcription as well as cell differentiation.
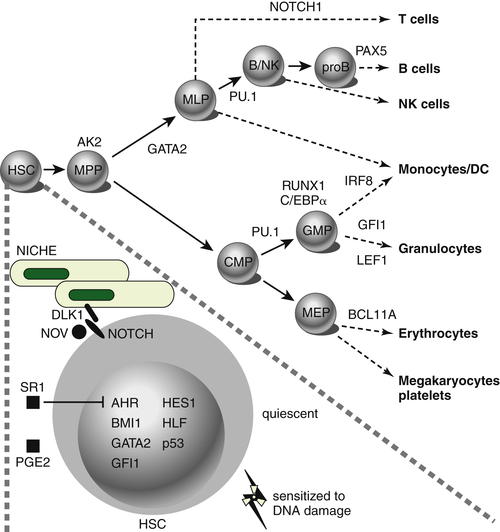
Figure 28-1 Transcription factors implicated in lineage specification of hematopoietic stem and progenitor cells. From Doulatov S et al. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120-136.
Table 28-2
Association of Karyotypic Aberrations with Molecular Findings in AML
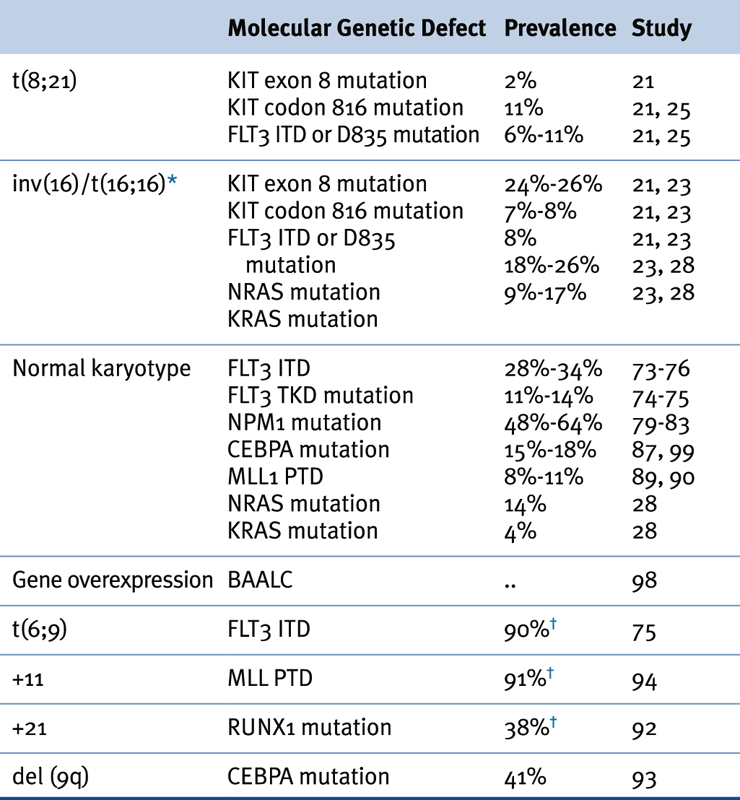
AML, Acute myeloid leukemia.
∗ ≤70% of inv(16) leukemias have mutations in receptor tyrosine kinase or RAS genes.
† Prevalence based on a limited number of cases.
Source: Adapted from Estey et al. Acute Myeloid Leukaemia. Lancet 2006;368:1894-1907, with permission.
The PML-RARα oncoprotein disrupts the interaction of retinoic acid and RARα, which converts the latter into a transcription activator, resulting in maturation arrest of hematopoietic progenitors at the promyelocyte stage. PML-RARα expression also disrupts PML localization, causing it to relocalize from discrete nuclear structures, the PML nuclear bodies, into microspeckled aberrant structures. PML-RARα acts as a dominant negative inhibitor of the PML protein, as well as the major heterodimeric partner of RARα, RXRα (retinoid X receptor). PML-RARα recruits several co-repressors, including the nuclear co-repressor (N-CoR). N-CoR inhibits transactivation from RARα target genes through the recruitment of the molecules sin3 and histone deacetylases, which in turn inhibit the binding of transcription factors and the binding of the transcriptional machinery to promoters, resulting in inhibition of gene expression for hematopoietic differentiation. Similarly, the PML moiety of the PML-RARα protein interacts with the DAXX co-repressor. Mutations preventing DAXX recruitment, although allowing PML-RARα dimerization, abrogated the ability of PML-RARα to block hematopoietic differentiation and immortalize cells. ATRA is the mainstay of therapy in APL, inducing leukemic cell differentiation and remission in patients with t(15;17)/PML-RARα or t(5;17)/NPM-RARα. Similarly, arsenic trioxide (As2O3) has been demonstrated to be effective in the treatment of de novo as well as of ATRA-resistant t(15;17)/PML-RARα APL. Current evidence suggests that the combination of ATRA and As2O3 represents a valid alternative to chemotherapy-containing regimens for the treatment of APL.
Core Binding Factor AMLs
CBF is a heterodimeric transcription factor that consists of a DNA binding α-subunit, encoded by one of three members of the RUNX family (RUNX1 or AML1, RUNX2, and RUNX3), and a β-subunit encoded by the CBFβ gene that increases DNA-binding affinity to the complex. Mutations involving CBF rearrangements occur in 15% of cases of AML and are associated with a favorable prognosis. CBF AML includes those carrying inv(16)/t(16;16), which gives rise to the fusion of CBFβ with the smooth muscle myosin heavy-chain gene (MYH11 or SMMHC), and t(8;21), which is associated with the fusion transcript composed of the AML1 and the eight-twenty-one (ETO) genes. Of note, in AML expressing the CBF translocation AML1-ETO, this fusion oncogene acts as a dominant negative inhibitor of native AML1. Similarly, the CBFβ-MYH11 oncoprotein is a dominant negative inhibitor of CBF both in transactivation assays and during development. In knockin CBFβ-MYH11 chimeric mice, CBFβ-MYH11 expression alters adult multilineage hematopoietic differentiation. CBFβ also modulates the effect of CBFβ-SMMHC in adult hematopoiesis and leukemogenesis.
Rearrangements of the MLL and HOX Genes
Approximately 4% of patients with de novo AML have balanced translocations or insertions involving the mixed-lineage leukemia (MLL) gene. MLL-gene fusions are highly associated with previous therapy that includes topoisomerase-II inhibitors. The estimated incidence of this type of rearrangement in secondary treatment-related AML is 2% to 12%. MLL is the human homologue of Drosophila TRX and constitutes a maintenance factor for HOX proteins, which are central during development and hematopoiesis. The N terminus of MLL, which contains the AT-hook DNA-binding motif and a region homologous to DNA methyltransferase, is always retained in the fusion protein arising from chromosomal translocations, whereas the C terminus, which contains the activation and SET domains, is always replaced by the fusion partner. The MLL AT hooks bind to the minor groove of DNA, which facilitates the binding and recruitment of transcription factors to promoter elements. 1 MLL fusion genes can initiate both myeloid and lymphoid leukemogenic programs depending on the fusion partner, of which more than 65 have been thus far identified in AML. Differential activation of the Wnt/β-Catenin pathway is required for the maintenance of MLL leukemia stem cells, and MLL-AF9, one of the most frequent fusion transcripts, requires interacting with the Polycomb Group protein CBX8 to induce a leukemogenic transcriptional program. Approximately 5% to 10% of AML cases present with rearrangements of MLL consisting of an in-frame partial tandem duplication (MLL-PTD) of exons 11-5 or 12-5. MLL-PTD promotes increased histone H3/H4 acetylation and methylation of H3 Lys4 at cis-regulatory HOXA sequences. Mislocalized activity of the H3K79 histone methyltransferase DOT1L has been proposed as a driver of leukemogenesis in AML carrying MLL rearrangements. Pharmacological inhibition of DOT1L with the selective DOT1L inhibitor EPZ004777 selectively inhibits H3K79 methylation and blocks expression of leukemogenic genes with little effect on non–MLL-translocated cells, suggesting that DOT1L inhibition represents a potential therapeutic option for patients with MLL rearrangements.
HOX genes are frequently overexpressed in leukemia. Constitutive HOX gene activation is required for MLL fusion protein-mediated AML. Gene expression profiling analysis showed that the HOXA4, HOXA9, HOXA10, PBX3, and MEIS1 homeobox genes are coexpressed across diverse cytogenetic groups but are undetectable in terminally differentiated hematopoietic cells. In AML, HOX genes are mainly disrupted via chromosomal translocation, such as the fusion of NUP98, NUP214 (also known as CAN), or MLL to HOXA9, HOXD13, DEK, and DDX10. Overexpression of HOXA6, HOXA7, HOXA9, and the HOX cofactor myeloid ecotropic viral integration site 1 (MEIS1) has also been correlated with chromosome 11q23 abnormalities involving the MLL protein, which directly regulates the expression of HOX genes. The caudal-type homeobox transcription factor 2 (CDX2) is overexpressed in 90% of patients with AML in spite of its lack of expression in hematopoietic progenitors. CDX2 overexpression in primary murine hematopoietic progenitors resulted in transplantable AML in vivo, which was associated with upregulation of HOXB6 expression, a protein that is overexpressed in 40% of cases of CN-AML. 2 This suggests the possibility that CDX2-mediated deregulation of HOX genes is a major pathway to leukemogenesis.
Mutations in the C/EBPα and PU.1 Genes
The C/EBPα gene, which encodes the CCAAT/enhancer-binding-protein-alpha, is a member of the family of leucine-zipper (bZIP) transcription factors that couples lineage commitment to terminal differentiation and cell cycle arrest in the process of myeloid differentiation. C/EBPα initiates growth arrest through induction of p21 and by disrupting the E2F transcriptional complexes during the G1 phase of the cell cycle. Mutations in the C/EBPα gene occur in 15% to 19% of patients with AML and normal cytogenetics. C/EBPα mutations increase the capacity of bone marrow myeloid progenitors to proliferate and predispose mice to a granulocytic myeloproliferative disorder. In the absence of specific C/EBPα mutations, decreased expression may serve as an alternative mechanism that disrupts C/EBPα gene function. For example, AML1-ETO appears to indirectly suppress C/EBPα expression by inhibiting positive autoregulation of the C/EBPα promoter. Notably, C/EBPα mutations, when biallelic, are associated with a favorable prognosis in patients with cytogenetically normal (CN)-AML.
The transcription factor PU.1 is indispensable for myelomonocytic differentiation during normal hematopoiesis and for regulating the commitment of multipotent hematopoietic progenitors. The course of AML in mice after knockdown of PU.1 includes a preleukemic stage during which immature myelomonocytic precursors accumulate in the bone marrow, followed by a leukemic phase with elevated leukemic blasts in peripheral blood. Also, PU.1-induced upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Despite its leukemogenic potential, PU.1 mutations have been rarely found in human AML.
Mutations Altering Signal Transduction
Mutations at several oncogenes promoting cell growth have been shown to participate in the pathogenesis of AML. FLT3 expression is restricted to CD34+ cells and a subset of dendritic precursors, where it regulates proliferation, differentiation, and apoptosis. 3 Internal tandem duplication (ITD) within the FLT3 juxtamembrane domain (exons 14 and 15) is among the most prevalent mutations in patients with CN-AML, being detected in about 30% of cases. Moreover, 7% of patients with AML harbor missense point mutations affecting the activation loop of the tyrosine kinase domain of FLT3 coded by exon 20, typically involving residue 835. There is evidence suggesting that FLT3 mutations occur in leukemic stem cells. In fact, 84% of patients with AML carrying FLT3-ITD mutations exhibit the same mutation at relapse. FLT3-ITD mutations activate aberrant signaling, including STAT5, PI3K/AKT, and MAPK pathways, and repress PU.1 and C/EBPα. FLT3 gene mutations are associated with high relapse rates and poor prognosis. Small-molecule tyrosine kinase inhibitors directed against the constitutively activated FLT3 protein such as midostaurin, sorafenib, or quizartinib have shown encouraging results in clinical trials. Further development of these agents is being pursued in the context of chemotherapy or hypomethylation-based combinatorial approaches.
Activation of the KIT tyrosine kinase by somatic mutation has been documented in a variety of human malignancies, including core binding factor AML, clustering within exon 17 and exon 8. KIT mutations confer a higher risk of relapse in patients with CBF AML. Gain-of-function KIT mutations may serve as a target for tyrosine kinase inhibitors (e.g., dasatinib), and their activity warrants further investigation in CBF AML harboring such mutations.
RAS oncogenes encode a family of membrane-associated proteins that regulate proliferation, differentiation, and apoptosis. The RAS proteins oscillate between a guanosine triphosphate (GTP)- and a guanosine diphosphate (GDP)-bound state. GDP-bound RAS is incapable of activating signal transduction pathways. NRAS mutations can be detected in 10% of patients with AML. Mutated RAS proteins are constitutively activated, which is held in their GTP-bound status, and efficiently induce an AML-like disorder in a mouse bone marrow transplantation model. The prognostic impact of RAS mutations in AML remains controversial. MEK inhibitors are currently being tested in patients with AML carrying RAS mutations.
Other Genetic Events Implicated in the Pathogenesis of AML
Mutations of the Nucleophosmin Gene
The NPM1 gene encodes a nucleus-cytoplasm shuttling protein implicated in preventing nucleolar protein aggregation, regulation of ribosomal protein assembly, initiation of centrosome duplication, and regulation of p53, p19ARF, and HDM2. NPM1 is involved in the control of primitive hematopoiesis, and mice lacking NPM1 alleles develop a syndrome reminiscent of human myelodysplasia. Translocations involving the NPM gene cause cytoplasmic dislocation of the NPM protein. NPM1 mutations are found in approximately 35% to 40% of adult patients with primary AML and are associated with high sensitivity to cytarabine-based therapy and to higher complete remission rates. Most of the cases (60% to 80%) carrying mutant NPM1 alleles correspond to patients with CN-AML. NPM1 mutations are typically heterozygous and almost exclusively map to exon 12. 4–6 A duplication of a TCTG tetranucleotide at position 956 to 959 accounts for 75% to 80% of cases. 6,7 Notably, NPM1 mutations are associated with a very favorable prognosis in the presence of wild-type FLT3 alleles, particularly when associated with IDH1 mutations. The recent development of experimental mouse models lacking NPM1 alleles or carrying NMP1 mutations will aid in understanding the role of NPM1 in the pathogenesis of AML and in developing novel targeted agents.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


