Fig. 37.1
Epidermal growth factor receptor structural and functional domains . (a) Schematic representation of EGFR sequence. L and CR, ligand-binding and cysteine-rich domains, TM transmembrane domain, TK tyrosine kinase domain, CT carboxyl-terminal domain. (b) Tyrosine-phosphorylation sites of EGFR and the bound signaling protein mediators.
The cytoplasmic domain of the receptor is characterized by a highly conserved tyrosine kinase (TK) domain. This region contains an ATP-binding pocket which is essential for the intrinsic TK activity of EGFR. The substrates of TK are several tyrosine residues next to the ATP-binding region or outside the ATP-binding pocket near the C-terminus of the receptor. Autophosphorylation of tyrosine residues induces conformational change of the protein, creating a docking structure for EGFR interaction with its downstream signaling mediators. In addition, seven serine and threonine residues bordering the TK domain are substrates for phosphorylation by non-receptor kinases, such as protein kinase C. Phosphoserine and phosphothreonine are also required for protein-protein interactions and have been inferred to be important for the receptor downregulation processes.
37.2.3 EGFR Ligands and Its Activatio n
EGFR activation is triggered by binding of its specific ligand. Since the discovery of EGF, several other ligands containing EGF-like domains have been identified as specific EGFR binding partners [9, 21, 22], including transforming factor α (TGF-α), amphiregulin, epiregulin, and β-cellulin. The soluble ectodomain of EGFR binds ligand, and this formation of a ligand-receptor complex on the cell surface induces the dimerization of EGFR [23, 24]. A 2:2 complex is formed in which the affinity of EGF or TGF-α to EGFR is 100–500 nM [25–27].
In the absence of ligand binding, EGFR exists on the cell surface as either monomer or dimer. However, ligand-induced dimerization is required for activation of the intrinsic TK activity [28–30]. Studies of the kinetics of stimulated and unstimulated EGFR have shown that ligand binding doubles the V max parameter and decreases the K m parameter for ATP by tenfold [31]. Binging to ligand increases the proportion of dimerized EGFR and reorientates the kinase domains in a way that increases the affinity for ATP binding due to conformational change, thus enhancing the enzymatic activity.
EGFR is autophosphorylated on several C-terminal tyrosines, including Tyr 992, Tyr 1045, Tyr 1068, Tyr 1086, Tyr 1148, and Tyr 1173 (Fig. 37.1b). The role of this autophosphorylation is to create a specific docking structure and facilitate the formation of protein complexes with EGFR downstream signal transduction partners. The formation of these protein complexes requires specific ternary configurations depending on autophosphorylation at specific tyrosine sites, leading to phosphorylation-dependent activation of major intracellular signaling pathways.
37.3 Biological Function and Related Signal Transduction Pathways of EGFR
Depending on the functional diversity of the proteins that EGFR complexes with, or is phosphorylated by, stimulation by EGFR ligands results in simultaneous activation of multiple signal transduction pathways (Fig. 37.2). Thus, activation of EGFR induces many biological activities, including evasion of apoptosis, promotion of cell growth, and cell migration. At the same time, ligand binding induces a rapid internalization of the receptors and enclaves them in endosome [32–34]. The internalized EGFR either undergoes degradation by locating to lysosomes or is recycled back to the cell membrane.
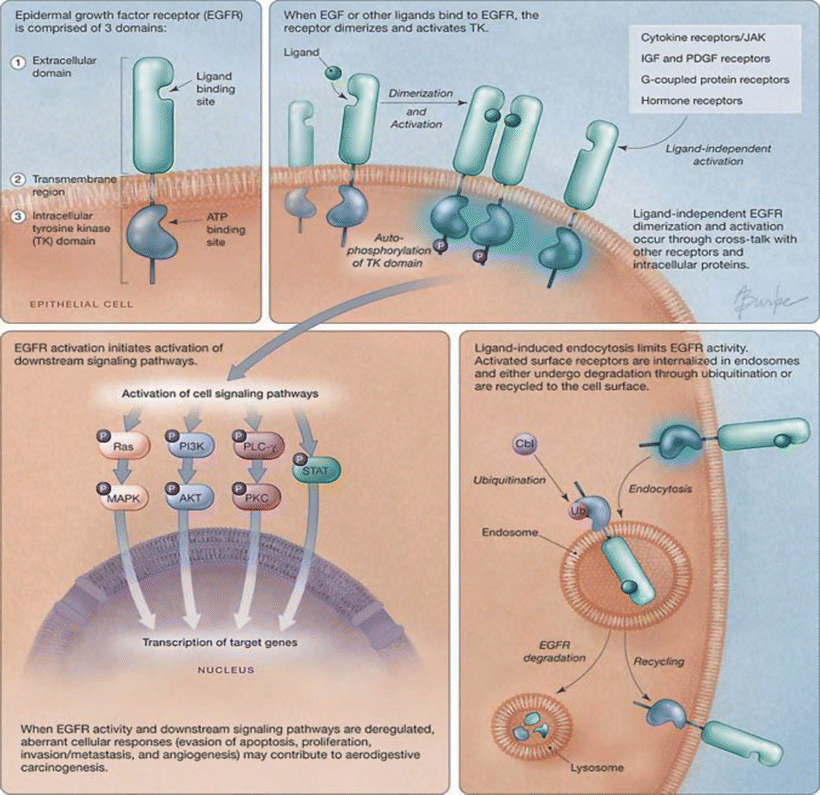
Fig. 37.2
Epidermal growth factor receptor activation, processing, and signaling . ATP adenosine triphosphate, Cbl a ubiquitin E3 ligase, IGF insulin-like growth factor, JAK Janus kinase, MAPK mitogen-activated protein kinase, P phosphate, PDGF platelet-derived growth factor, PI3K phosphatidylinositol-3-kinase, PKC protein kinase C, PLC phospholipase C, STAT signal transducers and activators of transcription, TK tyrosine kinase, Ub ubiquitin (adapted from Karamouzis, M. V. et al. JAMA 2007;298:70–82).
37.3.1 Ras/Mitogen-Activated Protein Kinase (MAPK) Pathway
RAS/MAPK is one of the major signaling pathways activated by EGFR. The formation of a protein complex between EGFR and Grb2 initiates signal transduction through the RAS/MAPK pathway . Grb can associate with EGFR either directly at Tyr 1068 and 1086 sites, or indirectly by binding to EGFR-associated Shc, which recognizes phosphorylated EGFR at Tyr1148 and 1173 [35, 36]. It can constitutively bind to Ras exchange factor Sos, resulting in the exchange of Ras-bound GDP for GTP and thus Ras activation. The activated Ras then activates the serine/threonine kinase Raf-1 [37, 38]. Activated Raf-1, through MEK, contributes to the phosphorylation, activation, and nuclear translocation of ERK1/2 (MAPK). ERK1/2 is responsible for phosphorylation of several nuclear transcription factors, such as c-fos and C/EBPs, which regulate cell proliferation and cell cycle progression [39, 40]. Activated MAPK also provides a negative feedback loop for this pathway by promoting dissociation of the Grb2/Sos complex and a reduction in the strength of signal transduction.
37.3.2 Phosphoinositide-3-Kinase (PI3K)/AKT Pathwa y
PI3K is a major regulatory protein that is involved in a variety of cellular activities, including cell proliferation, survival, adhesion, and migration [41–43]. Among its three classes, only Class Ia is activated by a tyrosine kinase receptor. One of the PI3K subunits, p85, can directly interact with Grb1/2 through its SH2 domain when Grb1/2 associates with EGFR at Tyr 1068 and Tyr 1086 [44]. Although the major binding partner of p85 is not EGFR, but ErbB3 [45, 46], activation of PI3K is found in response to the EGFR ligand through EGFR/ErbB3 heterodimers. Activated PI3K-Ia produces phosphatidylinositol-3,4,5-trisphosphate (PIP3). One of the well-known targets of this second messenger is the ser/thr kinase AKT [47]. Phosphorylated AKT can prevent apoptosis through targeted inhibition of Bad, a pro-apoptotic member of the Bcl-2 family and caspase 9, an enzyme involved in the Fas-mediated death pathway [48, 49]. PI3K/AKT also regulates the mammalian target of rapamycin (mTOR), which is responsible for the activation of translational machinery [50] and regulation of angiogenesis [51].
37.3.3 Phospholipase C-γ (PLCγ) and Protein Kinase C (PKC) Pathway s
EGFR signaling also affects cellular phospholipid metabolism. In addition to PI3K, it is found that PLCγ is regulated by EGF stimulation [52]. PLCγ binds directly to EGFR at Tyr 992 and 1173 and can be phosphorylated by EGFR at Tyr 771 and Tyr 174 [53]. The activated PLCγ catalyzes the hydrolysis of PtdIns [4, 5]-P2 to yield the important secondary messengers 1, 2-diacylglycerol (DAG) and inositol 1,3,5-triphosphate (IP3). IP3 mediates calcium release and at the same time affects the Ca2+-dependent enzyme PKC, which uses DAG as its cofactor. Thus, through PLCγ, EGFR can activate the Ca2+-dependent PKC pathway, which plays important roles in cell differentiation, proliferation, migration, and invasion [54, 55].
37.3.4 Janus Kinases (JAKs) and Signal Transducers and Activators of Transcription (STATs) Pathway s
STATs are identified as signal transducers upon activation by cytokine receptors [56, 57]. There are at least seven STAT genes that have been identified in mammals, including STAT-1 to STAT-4, STAT-5a, STAT-5b, and STAT6. Homo- and heterodimerization of STAT proteins allows them to translocate into the nucleus and work as transcription factors for activation of gene expression in response to cell growth and proliferation. STATs, particularly STAT-1, STAT-3, and STAT-5, have been implicated in EGFR signaling. However, their mode of activation is significantly different from that by cytokine receptor. First, the EGFR ligand-stimulated phosphorylation of STATs does not require cytokine receptor JAK kinases. Second, STATs do not bind to the C-terminal phosphotyrosines of EGFR; instead they are constitutively associated with EGFR [58, 59]. Activation of STAT transcription activity strictly depends on EGFR tyrosine kinase activity [60, 61]. Moreover, Src kinase may play a role in EGF-dependent STAT activation [59, 62].
37.3.5 Src Pathway
Src and its family members have been extensively reported to support EGFR activity [63–65]. Overexpression of Src proteins strongly enhances EGF-mediated proliferation and transformation of fibroblasts and epithelial cells [66, 67]. Src does not bind to the major autophosphorylation site of EGFR, but phosphorylates EGFR in its kinase domain at Tyr891 and 920 sites [68]. These two phosphotyrosines bind the SH2 domain of Src and may provide a docking site for the p85 subunit of PI3K. In addition, two other Src-dependent phosphorylation sites were identified at Tyr 845 and Tyr 1101 in the EGFR cytoplasmic domains [69]. Phosphorylated Tyr 1101 facilitates Src binding to EGFR, while phosphorylation of Tyr 845 is required for Src mediated STAT5b binding [62]. It is possible that Src serves as a signal transducer downstream of EGFR or a contributor to EGFR activation.
37.3.6 Transactivation of EGFR
In addition to ligand binding, EGFR can be functionally activated through cross-talk with other cell surface receptors. G-protein-coupled receptors [70, 71], such as prostaglandin E2 receptors and gastrin-releasing peptide receptor, platelet-derived growth factor receptor [72], insulin-like growth factor receptor [73], and some hormone receptors [74] also transactivate EGFR-mediated signal transduction pathways.
37.4 Oncogenic Activities and Expression of EGFR in Human Cancer Cell Lines and Tissues
37.4.1 In Vitro and In Vivo Studies of EGFR
The oncogenic activity of EGFR is evident from its ability to transform normal cells and promote cell proliferation. EGFR, by itself, is insufficient to cause transformation, but the addition of EGF, TGF-α, or other EGFR-specific ligands, or the existence of activation mutations is necessary for manifestation of the transformation phenotype [8, 75–77]. In addition to cell line studies, the use of transgenic animals has allowed the study of whether overexpression of EGFR or its ligands is indeed causative for hyperproliferation or tumor formation. Using TGF-α as an example, transgenic mice that overexpressed TGF-α driven by a keratin promoter show hypertrophy and hyperkeratosis accompanied by alopecia or stunted hair growth [78, 79]. TGF-α transgene expression is also linked to the appearance of papillomas following irritation or wounding. Targeted expression of TGF-α in the mammary gland results in hyperplasia, cystic expansion, and papillary adenoma following multiple pregnancies and lactation [80]. Therefore, overexpression of TGF-α is responsible for hyperproliferative responses but does not generally lead to tumors in rodents, suggesting that the TGF-α/EGFR signaling pathway provides the initial step in multistage carcinogenesis. Neoplastic transformation requires the overexpression or mutation of other proteins in the target tissues, such as c-Myc oncoprotein. This notion is supported by studies using mice with double transgenes of TGF-α and c-Myc that show development of mammary tumors [81].
Constitutively activated EGFR with no requirement for ligand binding can result from mutations that are found in many tumor tissues. A well-known example of a naturally occurring EGFR deletion mutant is EGFRvIII (Δ2-7) [82, 83], although other classes of EGFR ectodomain mutations EGFRvI and vII have also been discovered [84]. The EGFRvIII mutant has been detected in 40–50 % of grade VI glioblastomas [85], in up to 70 % of medulloblastomas and a small proportion of breast, ovarian, prostate, lung, and head and neck carcinomas [86–90]. Due to the deletion of exons 2–7, EGFRvIII lacks most of the extracellular domain, thus cannot be activated by ligand. Instead, it is constitutively activated and not internalized, resulting in constitutive long-term signaling which supports cell proliferation. A similar situation is observed in breast cancer, in which the Her2/neu or erbB2 oncogene was identified [91]. Her2/neu has a truncated extracellular domain and a point mutation in the transmembrane region, which activates the protein by inducing its dimerization in the absence of ligand. This receptor is a major oncogene and is overexpressed in 20–25 % of breast cancer tissues and associated with shortened survival [92].
Other activating mutations of EGFR are commonly found in lung cancers, and also observed in esophageal and head and neck cancers [93–96]. A deletion in exon 19 and nucleotide substitution in exon 21, such as L858R and L861Q, are the most common EGFR mutations in lung cancer cells. These genotypes have been identified as EGFR-TK inhibitor (TKI)-sensitizing mutations, because tumor cells harboring these mutations showed greater sensitivity to an EGFR-TKI (gefitinib) than those maintaining the wild-type gene [97, 98]. The mutations were suggested to stabilize the binding of EGFR with ATP or its competitive inhibitors. These mutations also induce anti-apoptotic pathways mediated by STAT-3 and AKT, strengthening the oncogenic activity of EGFR. However, a recent study using samples from the NCIC CTG trial showed that the presence of an EGFR mutation may increase responsiveness to EGFR-TKI, but is not indicative of a survival benefit [99].
37.4.2 EGFR Expression in Solid Malignancies
Since EGFR activation results in cell proliferation, inhibition of apoptosis, and the formation of neo-vasculature, several studies have evaluated the impact of EGFR activation on the clinical outcomes of patients with various solid malignancies. These include evaluation of ligand expression/concentrations, receptor expression, and activation of downstream signals such as MAP kinase.
EGF is a potent mitogen in the gastrointestinal tract and has been shown to increase proliferation in glandular cells in the colon [100]. TGF-α, another EGFR ligand, is also a growth factor in colonic epithelial cells [101]. Varying levels of receptor overexpression have been noted in colon cancer. Although overexpression of the receptor is noted in tumor tissues of approximately two-thirds of patients with colon cancer, the impact on prognosis is less certain [102–104]. EGFR expression may increase the propensity to develop hepatic metastasis [105]. EGFR is also frequently over expressed in aerodigestive malignancies. In patients with non-small cell lung carcinoma (NSCLC), EGFR overexpression is noted in approximately 40–80 % of the tumors [106–109]. In some studies, a higher level of EGFR expression has been linked to more advanced stages of disease at the time of diagnosis, and also to a poor prognosis [110–112]. More recently, aberrant activation of the EGFR pathway has been shown to result from mutations in the tyrosine kinase binding domain of EGFR [93]. Such mutations are noted in approximately 10–15 % of Caucasian patients. EGFR mutations are noted more frequently in women, patients with adenocarcinoma histology, those who have never smoked and in patients of Asian ethnicity [94]. High copy numbers of the EGFR gene also appear to have an impact on prognosis and may also predict heightened sensitivity to treatment with EGFR inhibitors [113, 114]. However, there does not appear to be a clear correlation between protein expression, gene copy number, or mutational status in NSCLC.
EGFR overexpression is also noted in a vast majority of tumor specimens from patients with head and neck carcinoma [115–117]. A higher level of EGFR expression in the tumor tissue relative to surrounding normal tissues has been noted by several investigators [118]. In many of these reports, EGFR overexpression was associated with an inferior clinical outcome and higher rate of disease recurrence following definitive local therapy. Aberrant activation of EGFR has also been noted in other solid malignancies such as breast, ovarian, pancreatic, and prostate cancers [102].
Based on reports of aberrant activation of EGFR in a variety of epithelial cancers, several agents that inhibit EGFR pathway activation have been tested in preclinical and clinical studies. Overexpression of the receptor in the tumor tissue relative to surrounding normal tissue provides the opportunity for cancer-selective therapy. Overexpression of EGFR and its ligands and the expression of constitutively active EGFR mutants support diverse cellular biological activities utilized by cancer cells to progress, including cell cycle progression, cell growth, differentiation, angiogenesis, invasion, and metastasis [9, 119, 120]. Elucidating the effects of EGFR on these biological activities has been greatly accelerated by the development of EGFR inhibitors in recent years.
37.5 Development of EGFR Inhibitors
The crucial roles that EGFR plays in tumor progression make this receptor an ideal target for the development of anti-cancer therapeutics [121–127]. There are several potential approaches to blocking EGFR signaling pathways. The receptor can be inhibited through its extracellular domain by antibodies and at the TK domain by small molecules [128]. In addition, antisense DNA/RNA , siRNA , or ribozymes can also be used to reduce expression of the receptor [129–131]. Table 37.1 summarizes EGFR-targeting agents that are currently being developed or are used in the clinic.
Table 37.1
EGFR inhibitors in clinical us e
Agent | Disease | Indication (FDA approved) |
|---|---|---|
Gefitinib | Non-small cell lung cancer | Treatment of advanced NSCLC following prior chemotherapy |
Erlotinib | Non-small cell lung cancer Pancreatic cancer | For advanced stage NSCLC, following 1 or 2 prior chemotherapy regimens In combination with gemcitabine for previously untreated advanced stage pancreatic cancer |
Lapatinib | Breast cancer | In combination with capecitabine for HER2-positive breast cancer |
Cetuximab | Colorectal cancer Head and neck cancer | For advanced stage colorectal cancer, in combination with irinotecan or as monotherapy (EGFR-positive tumors) In combination with radiation or as monotherapy |
Panitumumab | Colorectal cancer | Monotherapy following failure of prior chemotherapy for advanced stage colorectal cancer (EGFR-positive tumors) |
Many synthetic and semi-synthetic compounds have been identified as EGFR-selective TK inhibitor s [128, 132–134], such as gefitinib (ZD1839, Iressa) [135, 136], CI-1033 [137–139], and erlotinib (OSI-774, Tarceva) [140]. These compounds are orally bioavailable and work as competitive inhibitors in the ATP-binding domain of EGFR. Both in vitro and in vivo preclinical studies show that they have various effects on tumor cells expressing EGFR. EGFR-TKIs inhibit cell growth of several human tumor cell lines by blocking phosphorylation of EGFR, ERK, and AKT, which may result from G1 arrest of the cell cycle by reduction of cyclin D1 [141] and upregulation of cyclin-dependent kinase inhibitors p21 and p27kip1 [142, 143]. These inhibitors also downregulate the secreted growth factors TGF-α, basic fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF), which are important autocrine/paracrine factors stimulating both tumor cell growth and angiogenesis [144, 145]. Thus, it is no surprise that EGFR-TKIs can provide significant growth inhibition of human tumor xenografts in animal models [144, 146–148]. Particularly, they markedly enhance the efficacy of cytotoxic agents including cisplatin, paclitaxel, and other platinums in the treatment of human tumor xenografts in nude mice [149–151]. Furthermore, EGFR-TKIs show synergistic inhibitory effects when they are combined with other targeted agents [152, 153]. For example, studies from our group and the others illustrate that the combination of EGFR-TKIs with cyclooxygenase-2 inhibitors (COX-2Is) either additively or synergistically reduces the growth rate of several types of cancer cells [154–157]. In addition, EGFR-TKIs have been combined with inhibitors of IGF-1R [158–160], mTOR [161, 162], and VEGF/VEGF-Receptor [144, 163–165], and antisense oligonucleotides against Bcl-2 [166] and MDM2 [167]. Simultaneously blocking the expression and/or activity of both EGFR and these proteins results in effective induction of cell cycle regression and apoptosis in vitro and in vivo.
Monoclonal antibodies (mAbs) directed against the extracellular domain of EGFR are another class of therapeutic agents that has been well-studied in recent years. Among the available anti-EGFR mAbs, the one currently used in the clinic is the chimeric human:murine mAb cetuximab (IMC-225, Erbitux) [168]. This antibody binds to EGFR with high affinity (K d = 0.39 nM), thereby competing with EGFR ligand binding and preventing ligand-induced EGFR activation [169–172]. In addition, cetuximab and other anti-EGFR antibodies can induce EGFR dimerization, resulting in receptor internalization and downregulation [173, 174]. Cetuximab also induces G1 arrest because of elevated levels of p27kip1 [175], followed by apoptosis [176]. Blockade of EGFR by cetuximab decreases tumor cell production of angiogenic factors, such as bFGF, VEGF, and interleukin-8 [177, 178], leading to a significant reduction in microvessel density and enhancement of apoptotic endothelial cells in human xenograft tumors. Cetuximab and similar anti-EGFR mAbs also inhibit the expression and activity of several matrix metalloproteinases (MMPs). The antibody-mediated reduction of MMPs results in inhibition of tumor cell invasion in vitro and metastasis in vivo [178–180]. Similar to EGFR-TKIs, cetuximab markedly augments the antitumor effects of chemotherapeutic agents such as cisplatin and doxorubicin in human xenograft tumor models [181, 182]. However, the functions of EGFR-TKIs and anti-EGFR mAbs are not completely overlapping. Anti-EGFR antibodies, but not EGFR-TKIs, have the ability to induce receptor internalization, an important mechanism for attenuating receptor signaling. Accordingly, the combination of an EGFR-TKI with cetuximab demonstrated a synergistic inhibitory effect on tumor cell growth in an in vitro study [183].
Recently, high-throughput screening approaches and rational drug design have been used to develop new EGFR inhibitors. These methods allow the identification of novel EGFR inhibitors that can be further modified chemically to target a tumor population that is resistant to current EGFR-targeting agents [184]. Structure-based rational drug design utilizes nucleic acid-directed gene silencing molecules including antisense oligodeoxynucleotides , RNA interference , and ribozymes to prevent EGFR translation from its mRNA [185]. Although these approaches are still in the early phase of development, they have great potential to enhance our ability to block EGFR and its related signaling pathways in a clinically applicable manner.
37.6 Clinical Application of EGFR Inhibitors
37.6.1 Non-small Cell Lung Cancer
EGFR inhibitors are now in routine clinical use for the treatment of advanced stage NSCLC . Gefitinib was the first drug among this class of agents to demonstrate anti-cancer activity in NSCLC. Two doses of gefitinib (250 and 500 mg/day) were compared to each other in patients with advanced stage NSCLC following progression with standard therapy [186, 187]. The objective response rate was 10–19 % and an additional 30–40 % of the patients experienced disease stabilization. Improvements in quality of life and lung cancer symptom scores were also noted in many patients. No difference in efficacy was noted between the two doses, though the lower dose was associated with a more favorable tolerability profile. This led to the approval of gefitinib for the treatment of advanced NSCLC by the Food and Drug Administration (FDA). However, a subsequent phase III study that compared gefitinib with placebo failed to demonstrate a survival advantage when gefitinib was used in second or third line treatment settings for advanced NSCLC [188]. Despite a response rate of 9 %, and modest disease stabilization rate, the lack of improvement in progression-free survival and overall survival with gefitinib effectively halted the use of this agent for the treatment of advanced stage NSCLC. However, there were selected sub-groups of patients, including never-smokers and those of Asian ethnicity, who did experience superior survival with gefitinib over placebo. This is suggestive of a role for this agent in clinically-selected or molecularly-selected subsets of patients with NSCLC.
Erlotinib, another EGFR-TKI, is also FDA approved for the treatment of advanced NSCLC. Following the demonstration of its anti-cancer activity in a phase II study [189], erlotinib was compared with placebo in a randomized phase III study (2:1 randomization) for advanced NSCLC patients. Those who progressed following one or two prior chemotherapy regimens were eligible for the study [190]. The median survival was superior with erlotinib (6.7 months vs. 4.7 months). The response rate was 9 %, and 35 % of patients experienced disease stabilization with erlotinib. Improvements were also noted in time to symptomatic deterioration for patients treated with erlotinib. The main side effects were skin rash (75 %) and diarrhea (54 %). The data from this study led to the approval of erlotinib for use as either second line or third line therapy of advanced stage NSCLC.
Cetuximab, a monoclonal antibody against EGFR also has efficacy as monotherapy, although the activity appears to be relatively modest. In a phase II study for patients with previously treated advanced NSCLC, cetuximab therapy demonstrated a response rate of 5 % and disease stabilization rate of 30 % [191]. The incidence of skin rash was more frequent, but diarrhea was uncommon with the antibody.
Following the demonstration of efficacy of EGFR inhibitors as monotherapies, randomized clinical trials have been conducted to study them in combination with chemotherapy. This approach was supported by preclinical data that demonstrated supra-additive effects when an EGFR inhibitor was co-administered with chemotherapeutic agents in NSCLC cell lines. However, there was no improvement in overall survival and progression-free survival for the combination of an EGFR-TKI and standard platinum-based chemotherapy over that with chemotherapy alone in patients with advanced stage NSCLC [192, 193]. Although the reasons behind the conflicting pre-clinical and clinical findings are not entirely clear, further development of combination regimens with EGFR-TKIs and chemotherapy has been discontinued.
On the contrary, the monoclonal antibody cetuximab was associated with promising response rates and survival duration when combined with chemotherapy in phase II studies for patients with advanced stage NSCLC [194–196]. This prompted phase III studies of chemotherapy with or without cetuximab in patients with chemotherapy-naïve advanced stage NSCLC. The results have been conflicting, with one study that combined cetuximab with chemotherapy (carboplatin and a taxane) showing no improvement in progression-free survival over chemotherapy alone, whereas another study demonstrated improved survival in patients with EGFR-expressing tumors who were treated with cetuximab in combination with chemotherapy (cisplatin and vinorelbine) (Press release, Imclone Pharmaceuticals, Inc., September 2007). The detailed results of this trial are yet to be reported. Of note, the positive study with cetuximab was unique in selecting patients for therapy based on EGFR expression status.
The clinical development of panitumumab, a fully human monoclonal antibody against EGFR, was discontinued based on data from a randomized phase II study (carboplatin and paclitaxel administered with or without panitumumab) [197]. There was no improvement in any of the efficacy parameters evaluated in the study with the addition of panitumumab to chemotherapy. Presently, the reasons behind the differential interaction of EGFR-TKIs and monoclonal antibodies with chemotherapy are unclear. It is conceivable that antibody-mediated cellular cytotoxicity (ADCC) might play a role. Furthermore, it can be hypothesized that the non-target effects of EGFR-TKIs may be responsible for their unfavorable interaction with chemotherapy.
EGFR inhibitors are now being evaluated for the treatment of earlier stages of NSCLC. This includes combination approaches with external beam radiation for locally advanced, surgically unresectable NSCLC and in the adjuvant therapy setting for patients with surgically-resected early stage NSCLC. Appropriate patient selection, using molecular and clinical factors is a key focus of ongoing research efforts.
37.6.2 Head and Neck Cancer
Several studies have documented the efficacy of EGFR inhibitors for the treatment of head and neck cancer. Both gefitinib and erlotinib have demonstrated single agent activity in this setting. Administration of gefitinib to patients with advanced head and neck cancer following progression with one prior chemotherapy regimen was associated with a response rate of 11 % and an overall disease control rate of 53 % [198]. In a small subset of patients who underwent sequential tumor biopsies, no consistent correlation was noted between response to therapy and changes in various biomarkers. Erlotinib was also associated with objective tumor responses in patients with advanced head and neck cancer [199]. Some recent studies have evaluated the combination of gefitinib or erlotinib with chemotherapeutic agents. A phase I/II study of erlotinib with cisplatin found response and disease stabilization rates of 21 and 49 %, respectively, in previously untreated patients with advanced head and neck cancer [200]. In another study that evaluated the combination of erlotinib, cisplatin, and docetaxel, a high response rate of 67 % was noted [201].
The monoclonal antibody cetuximab has received FDA approval for the treatment of patients with locally advanced head and neck cancer, either as a single agent in patients with advanced platinum-refractory disease, or in combination with radiation for localized disease. Since EGFR activation is a survival pathway for irradiated cells [202], the combination of radiotherapy with an EGFR inhibitor was evaluated in a randomized phase III study in patients with locoregional head and neck cancer [203]. Concurrent administration of external beam radiation with cetuximab resulted in near doubling of the median survival over that with radiation alone. The median duration of local control was 24 months with cetuximab and radiation compared to 15 months with radiotherapy alone. Notably, there was no increase in radiation-related toxicities with the addition of cetuximab, with the exception of skin rash. These results led to the evaluation of cetuximab in combination with radiation in the treatment of other solid malignancies.
When given to patients with advanced head and neck cancers as monotherapy following failure with prior platinum-based therapy, cetuximab was associated with a response rate of 13 % and a disease stabilization rate of 46 % [204]. Cetuximab has also been shown to overcome resistance to platinum-based chemotherapy . The administration of cetuximab in combination with cisplatin to patients who failed prior platinum-based therapy resulted in a response rate of 10 % and disease control rate of 53 % [205]. The favorable tolerability profile and anti-cancer activity associated with the combination of cetuximab and platinum-based chemotherapy was further substantiated by a phase II study by Herbst et al. [206]. These findings were confirmed by a phase III study, which demonstrated a superior response rate with cetuximab and cisplatin when compared with cisplatin alone (26 % vs. 10 %) and a non-significant trend towards improved progression-free survival with the combination (4.2 months vs. 2.7 months) [207]. Another phase III trial conducted recently confirmed the utility of cetuximab in first line therapy of head and neck cancers. The median survival associated with cetuximab in combination with platinum-5-fluorouracil was superior to that of chemotherapy alone in patients with recurrent or metastatic squamous cell carcinoma of the head and neck (10.1 months vs. 7.4 months) [208]. EGFR inhibitors are now being evaluated in combination with other targeted agents and also in earlier stages of head and neck cancer as adjuvant or neo-adjuvant therapy.
37.6.3 Colorectal Cancer
Cetuximab is efficacious in the treatment of advanced colorectal cancer, both as a monotherapy and in combination with chemotherapy. In a large phase II study (n = 346 patients), cetuximab was administered as a monotherapy to patients who had progressed with standard chemotherapeutic regimens [209]. Expression of EGFR in the tumor (immunohistochemistry positive) was mandated as an entry criterion. The response rate (12 %) and median overall survival (6.6 months) were suggestive of single agent activity for cetuximab in this setting. Subsequently, a phase III study compared cetuximab with best supportive care in patients with EGFR-expressing colorectal cancer, following progression with standard chemotherapy [210]. The response rate was 8 % with cetuximab and there was a significant improvement in progression-free survival and overall survival. Patients treated with cetuximab reported less deterioration in global health status at 8 weeks and 16 weeks of therapy, compared to that with supportive care alone.
Cetuximab has also undergone extensive evaluation in combination with chemotherapy for the treatment of advanced colorectal cancer. In a study for irinotecan-refractory colorectal cancer, patients were randomized to treatment with cetuximab alone or in combination with irinotecan [211]. The combination was associated with a higher response rate than that with monotherapy (23 % vs. 11 %). The median time to progression (4.1 months vs. 1.5 months) and overall survival (8.6 months vs. 6.9 months) were also superior for the combination. Interestingly, anti-cancer activity was also noted when irinotecan was added to cetuximab after progression with monotherapy. This was consistent with preclinical studies that demonstrated reversal of resistance to irinotecan by the addition of cetuximab [212]. This notion was further substantiated by a non-randomized study (n = 1123 patients) that treated irinotecan-refractory, EGFR-expressing colorectal cancer patients with the combination of cetuximab and irinotecan [213]. The median progression-free survival rate of 34 % at 24 weeks met the primary endpoint of the study.
Recent studies have focused on the evaluation of cetuximab in patients who had not received any prior chemotherapy for advanced colorectal cancer. A large phase III study randomized EGFR-expressing advanced colorectal cancer patients to treatment with 5-fluorouracil, leucovorin, and irinotecan (FOLFIRI) with or without cetuximab [214]. The addition of cetuximab to chemotherapy resulted in a superior response rate (47 % vs. 39 %) and a modest improvement in median progression-free survival (8.9 months vs. 8 months). Cetuximab also improved the efficacy of oxaliplatin-based chemotherapy regimens. In a randomized phase II study, the addition of cetuximab to 5-fluorouracil, leucovorin, and oxaliplatin (FOLFOX) resulted in a higher response rate than with FOLFOX alone (46 % vs. 37 %) [215]. These studies suggest a role for cetuximab in combination with chemotherapy as front-line therapy for advanced colorectal cancer. Cetuximab also appears to enhance the efficacy of other molecularly-targeted agents. Bevacizumab, a monoclonal antibody against the vascular endothelial growth factor, is commonly used in combination with standard chemotherapy for advanced colorectal cancer. In a randomized phase II study, the combination of cetuximab and bevacizumab resulted in a response rate of 23 % in patients with irinotecan-refractory colorectal cancer [216]. Thus, in addition to its routine use in patient care, cetuximab is being extensively studied in various settings for the treatment of colorectal cancer.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


