Biological hallmarks of cancer
Douglas Hanahan, PhD  Robert A. Weinberg, PhD
Robert A. Weinberg, PhD
Overview
An enigma for cancer medicine lies in its complexity and variability, at all levels of consideration. The hallmarks of cancer constitute an organizing principle that provides a conceptual basis for distilling the complexity of this disease in order to better understand it in its diverse presentations. This conceptualization involves eight biological capabilities—the hallmarks of cancer—acquired by cancer cells during the long process of tumor development and malignant progression. Two characteristic traits of cancer cells facilitate the acquisition of these functional capabilities. The eight distinct hallmarks consist of sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, deregulating cellular energetics and metabolism, and avoiding immune destruction. The principal facilitators of their acquisition are genome instability with consequent gene mutation and tumor-promoting inflammation. The integration of these hallmark capabilities involves heterotypic interactions among multiple cell types populating the “tumor microenvironment” (TME), which is composed of cancer cells and a tumor-associated stroma, including three prominent classes of recruited support cells—angiogenic vascular cells (AVC), various subtypes of fibroblasts, and infiltrating immune cells (IIC). In addition, the neoplastic cells populating individual tumors are themselves typically heterogeneous, in that cancer cells can assume a variety of distinctive phenotypic states and undergo genetic diversification during tumor progression. Accordingly, the hallmarks of cancer—this set of necessarily acquired capabilities and their facilitators—constitute a useful heuristic tool for elucidating mechanistic bases and commonalties underlying the pathogenesis of diverse forms of human cancer, with potential applications to cancer therapy.
Distilling the dauntingly complex manifestations of cancer
As outlined in the preceding chapter, and comprehensively described elsewhere in this encyclopedic textbook, the manifestations of cancer are disconcertingly complex and diverse. Cancers affecting different organs vary dramatically, in regard to genetics, histopathology, effects on systemic physiology, prognosis, and response to therapeutic intervention, explaining why the discipline of oncology is largely balkanized into organ-specific specialties, and why the chapters of this textbook are largely aligned as individualistic descriptions of organ-specific cancers.
In the face of this disconcerting diversity and complexity of disease manifestations, one might ask whether there are underlying principles—mechanistic commonalities—masked by the genetic and phenotypic complexities that span the multitude of cancer types and forms. In 2000, and again in 2011, we put forward a hypothesis that the vast complexity of human cancers reflects different solutions to the same set of challenges, namely that the lesions we observe in the forms of symptomatic neoplastic disease have all necessarily acquired, by various strategies, a common set of distinct functional capabilities that enable inappropriately chronic cell proliferation, and the focal or disseminated growth of populations of neoplastic cancer cells. We proposed to call this set of acquired capabilities “hallmarks of cancer.”1, 2 We further suggested that two characteristic traits of neoplastic growths—elevated mutability of cancer-cell genomes and inflammation by complex arrays of immune cells—are the key facilitators used by incipient neoplasias to acquire essential hallmark capabilities. Our current conceptualization of the biological hallmarks of cancer incorporates the eight distinct functional capabilities and the two enabling facilitators, these being schematized in Figure 1.
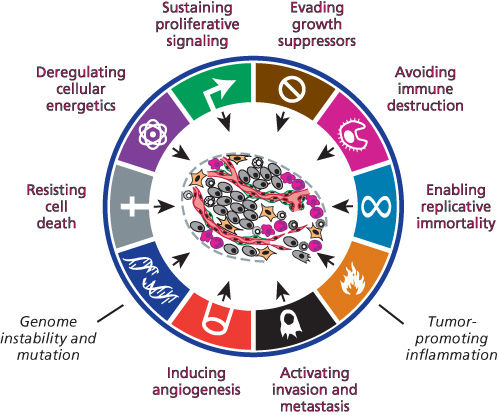
Figure 1 The biological hallmarks of cancer. The schematic illustrates what are arguably necessary conditions to manifest malignant disease—the hallmarks of cancer—comprising eight distinct and complementary functional capabilities and two facilitators (in black italics) of their acquisition.1, 2 These halllmark traits may be acquired at different stages in the multistep development of cancer, via markedy distinctive mechanisms in different forms of human cancer. Two aberrant characteristics of cancerous lesions are demonstrably involved in facilitating the acquisition during tumorigenesis of these functional capabilities: genome instability and the resultant mutation of regulatory genes, and the infiltration of immune inflammatory cells endowed by their biology—for example involvement in wound healing—to contribute to one or another hallmark capability. Different forms of cancer may be more or less dependent on a particular hallmark. Thus, adenomatous tumors typically lack the capability for invasion and metastasis. Leukemias may not require angiogenesis or invasive ability, although progression to lymphoma almost certainly requires both. The necessity of evading tumor immunity may be less important for certain cancers but is increasingly appreciated to be widespread.
The following sections describe these 10 key aspects of cancer pathophysiology. Then we introduce the observation that cancer cells recruit a variety of normal cell types that contribute in various ways the acquisition of hallmark functionalities. We conclude with a brief discussion on potential clinical implications of the hallmarks concept. For further detail and background, the reader is referred to our initial publications laying out the concept of the hallmarks of cancer,1, 2 as well as to another perspective that expands on the roles of stromal cells in enabling the hallmarks of cancer.3 Notably, only a few recent publications not cited in these three perspective articles are referenced herein. A textbook on the biology of cancer4 may provide additional details on many of the mechanisms of cancer pathogenesis described in outline in this chapter.
Acquired functional capabilities embody biological hallmarks of cancer
In our current conceptualization, there are eight hallmark capabilities that are common to many, if not most forms of human cancer (Figure 1). Each capability serves a distinct functional role in supporting the development, progression, and persistence of tumors and their constituent cells, as summarized briefly in the following sections.
Hallmark 1: sustaining proliferative signaling
The defining criterion of cancer as a disease is chronic, inappropriate cell proliferation, which results from corruption of cellular regulatory networks that normally orchestrate (transitory) proliferation of cells during embryonic development, physiological growth, and homeostatic maintenance of tissues throughout the body. Both positive (inductive) and negative (repressive) signals govern cell division and proliferation. Thus, this first hallmark capability embodies a complex set of inductive signals that instruct entry into and progression through the cell growth-and-division cycle to produce daughter cells. In the context of cancer, such stimulatory signals are activated and, in contrast to normal situations in which proliferative signaling is transitory, the signals are sustained chronically.
The most well-established and widespread mechanism of sustaining proliferative signaling involves mutational alteration of genes within cancer cells that convert such genes into active drivers of cell proliferation. These activated genes—defined as oncogenes—render otherwise transitory proliferation-promoting signals chronic. Such oncogenes typically encode proteins altered in structure and function or abundance compared to their normal cellular counterparts, which are responsible for receiving proliferative signals from extracellular sources and transmitting the signals through complex regulatory circuits operating within the cell.
Prominent examples of mutated driver oncogenes that sustain proliferative signaling in human cancers include the epidermal growth factor (EGF) receptor and signal transducers in the downstream KRAS–RAF–MEK–MAPK pathway that process and transmit growth-stimulatory signals via a succession of protein phosphorylations to the cell-division machinery operating in the nucleus. Mutations that render one or another of these proteins chronically active are found in many forms of human cancer, including the aforementioned EGFR and related receptor tyrosine kinases such as HER2 and ALK; similarly acting mutations result in chronic activation of the downstream signal transducers KRAS, BRAF, and MEK. We note, however, that activation in cancer cells of this central mitogenic pathway does not invariably depend on genetic changes acquired during the course of tumor progression. In certain instances, epigenetic deregulation of autocrine (autostimulatory) and paracrine (cell-to-cell) signaling circuits can also provide cancer cells with chronic growth-promoting signals, doing so in the apparent absence of underlying somatic mutations.
Hallmark 2: evading growth suppressors
The essential counterbalance to proliferative signals in normal cells are braking mechanisms that either overrule the initiation of, or subsequently block, the cell-division process instigated by such signals. The genes encoding these proteins are often termed tumor suppressor genes (TSGs). The most prominent brakes are the direct regulators of progression through the cell growth-and-division cycle, embodied in the retinoblastoma protein (pRb) and several “cyclin-dependent” kinase-inhibitor proteins. The activity of this molecular-braking system is itself normally regulated by the integration of extracellular pro- and antigrowth signals transduced by receptors on the cell surface, along with monitors of the intracellular physiologic state of the cell, in order to regulate tissue homeostasis and orchestrate transitory physiological proliferation.
An intracellular monitoring system, which is centered upon the p53 protein, serves to ensure that cells only advance through their growth-and-division cycles when the physiologic state of the cell is appropriate. Thus, p53 detects unrepaired damage to a cell’s genome as well as stressful physiologic imbalances that could impair accurate genome duplication, chromosomal segregation, and cell division. In response to cellular stress alarms, p53 then proceeds to activate inhibitors of the cell-cycle machinery. In cases of severe genomic damage or stressful physiological abnormalities, p53 and its associates can instead induce programmed cell death (see below), an extreme form of putting on the brakes to cell proliferation.
A number of component genes in both of these generic braking mechanisms—the Rb and p53 pathways—are classified as TSGs by virtue of their frequent loss-of-function via deletion or intragenic mutations; alternatively, other mechanisms may achieve the same end by shutting down expression of these genes through epigenetic mechanisms, notably those involving DNA and histone methylation. Thus the p53 gene is mutated in ∼40% of all human cancers, and many of the remaining tumors with wild-type p53 instead carry genetic lesions or epigenetic alterations that compromise p53 signaling in other ways.
Genetic profiling of genomes and transcriptomes indicates that a majority of human tumors contain defects—genetic or epigenetic—in the functions of the Rb and p53 tumor-suppressor pathways. Moreover, a large body of functional studies involving manipulation of these pathways in cultured cancer cells and mouse models of tumor initiation, growth, and malignant progression have clearly established the critical importance of TSGs in these pathways as significant barriers to the development of cancer. As such, evasion of growth suppressors is clearly a hallmark capability, necessary to ensure that continuing cancer cell proliferation and consequent tumor growth is not halted by braking mechanisms that, under normal circumstances, limit the extent of cell proliferation in order to maintain tissue homeostasis.
Hallmark 3: resisting cell death
There exists a second, fundamentally distinct barrier to aberrant cell proliferation, which involves intrinsic cellular mechanisms that can orchestrate the programmed death of cells deemed to be either aberrant or, in the case of normal development and homeostasis, superfluous. The most prominent form of programmed cell death is apoptosis, the genetically programmed fragmentation of a cell destined to die. Included among the situations where normal cells activate their apoptotic program to die are ones where the cell is damaged in various ways, mislocalized, or inappropriately migrating or proliferating. The apoptotic program can be triggered by cell intrinsic and non-cell-autonomous signals that detect different forms of cellular abnormality.
The apoptotic cell-death program involves the directed degradation of the chromosomes and other critical cellular organelles by specialized enzymes (e.g., caspases), the shriveling and fragmentation of the cell, and its engulfment, either by its neighboring cells or by tissue-surveying phagocytes, notably macrophages. The apoptotic cascade is completed in less than an hour in mammalian tissues, explaining why apoptotic cells are often surprisingly rare when visualized in tissue sections, even in a population of cells experiencing apoptosis-inducing environmental conditions, such as cancer cells in tumors subjected to cytotoxic chemotherapy or to acute hypoxia consequent to vascular insufficiency.
The rapid engulfment of apoptotic cell bodies ensures that their death does not release subcellular components that would otherwise provoke an immune response; this “immune silence” contrasts with a second form of programmed cell death: necroptosis. Long known as necrosis and envisioned as the passive dissolution of a dying cell, necrosis can also be an active, programmed process that is governed by cellular regulators and effectors distinct from those regulating apoptosis. Necroptosis can be activated by various conditions, including oxygen and energy deprivation, viral infection, and inflammation.5 Cells dying by necroptosis (or passive necrosis) rupture, releasing their contents and leaving their carcasses as immunogenic debris that can attract (or exacerbate) an immune inflammatory response, which, as discussed below, can have both tumor-promoting and tumor-antagonizing effects.
A third program capable of inducing cell death, termed autophagy, serves as a recycling system for cellular organelles that can help cells respond to conditions of nutrient deprivation, by degrading nonessential cellular organelles and recycling their component parts. Thus, autophagy generates metabolites and nutrients necessary for survival and growth that cells may be unable to acquire from their surroundings. In addition, while generally a survival system, extreme nutrient deprivation or other acute cellular stresses can lead to a hyperactivation of autophagic recycling that drives a cell to a point-of-no return, in which its complement of organelles falls below the minimum level required for viability; as a consequence, the cell dies via “autophagy-associated” cell death, distinct in its characteristics from both apoptosis and necroptosis. Stated differently, depending on the physiologic state of a neoplastic cell, autophagy may either sustain its survival and facilitate further proliferation or eliminate it via autophagy-associated cell death.6
These three distinct mechanisms for triggering cell death must be variably circumvented or attenuated by cancer cells if they and their descendants are to continue their proliferative expansion and phenotypic evolution to states of heightened malignancy.
Hallmark 4: enabling replicative immortality
A third intrinsic barrier to chronic proliferation is integral to the linear structure of mammalian chromosomes: the telomeres at the ends of chromosomes record—by progressive reduction of their length during each cell-division cycle—the number of successive cell generations through which a cell lineage has passed. The telomeres are composed of thousands of tandem copies of a specific hexanucleotide DNA sequence located at the ends of every chromosome that are associated with a specialized set of DNA-binding proteins. Operating together, these nucleoprotein complexes protect the ends of chromosomes both from degradation by the DNA-repair machinery, which is designed to detect DNA damage, and from end-to-end fusions with other chromosomes catalyzed by naked DNA ends.
Notably, when the number of telomere repeats erodes below a certain threshold, a tripwire is triggered, causing cell-cycle arrest or apoptosis mediated by the p53 tumor-suppressor protein, operating in its role to sense DNA damage. Circumventing these p53-induced antiproliferative responses (e.g., by mutational inactivation of the p53 gene) allows cancer cells with eroding telomeres to ignore the short-telomere checkpoint and continue proliferating, but only transiently. Sooner or later, the continuing erosion of telomeric DNA leads to loss of the protective nucleoprotein caps protecting the chromosomal DNA ends, which allows end-to-end fusions of chromosomes, breakage–fusion–bridge cycles during mitosis, and resultant karyotypic chaos that leads to cell death instead of cell division.
The cancer cells in many fully developed tumors circumvent the proliferative barrier presented by telomere erosion and the imminent mitotic catastrophe of telomere dysfunction by activating a system for telomere maintenance and extension that is normally used to preserve the replicative capacity of normal embryonic and tissue stem cells. This system involves expression of the telomere-extending enzyme named telomerase. Less frequently, they engage an alternative interchromosomal recombination-based mechanism for preserving telomere length. Thus, through one strategy or another, cancer cells acquire the capability to maintain their telomeres, avoiding the barrier of intolerably shortened telomeres, thereby enabling the unlimited replicative potential—termed cellular immortality—that is required for continuing expansion of populations of cancer cells.
Hallmark 5: inducing angiogenesis
Like normal organs, tumors require a steady supply of oxygen, glucose, and other nutrients, as well as a means to evacuate metabolic wastes, in order to sustain cell viability and proliferation. The tumor-associated vasculature serves these purposes. The deleterious effect that ischemia has in normal tissue is well established clinically and experimentally: cells die, via one form of programmed cell death or another, causing tissue and organ degradation and dysfunction. Similarly, the growth of developing nests of cancer cells halts when their ability to acquire blood-borne nutrients becomes inadequate, typically when the nearest capillary is more than 200 µ away. Angiogenesis—the formation of new blood vessels—is commonly activated and demonstrably beneficial for many tumor types.
Cells at the diffusion limit from the nearest capillary activate various stress-response systems, of which the most prominent involves the hypoxia-inducible transcription factors (HIF), which regulate hundreds of genes, including ones that directly or indirectly induce angiogenesis and other stress-adaptive capabilities. Much like cells in ischemic tissues, cancer cells lacking sufficient oxygen and glucose will typically die by necrosis/necroptosis, apoptosis, or rampant autophagy. This explains why most vigorously growing tumors are well vascularized with evidence of ongoing angiogenesis.
Of note, the tumor-associated neovasculature is usually aberrant both morphologically and functionally. Tumor blood vessels are tortuous, dilated, and leaky, with erratic flow patterns and “dead zones” in which no blood flow is detectable, in marked contrast to the seamless blood flow operating in the normal vasculature. Moreover, the degree of vascularity varies widely from one tumor type to another, ranging from intensely vascularized renal carcinomas to poorly vascularized pancreatic ductal adenocarcinomas.
Finally, we note that while chronic angiogenesis is a hallmark of most solid tumors, some may devise an alternative means to acquire access to the vasculature: in certain cases, cancers coopt normal tissue vasculature, by employing the hallmark capability for invasion and metastasis. Thus, particular types of cancer cells can proliferate and grow along normal tissue capillaries, creating sleeves whose outer diameters are dictated by the 200-μ diffusion limit. While vascular cooption is evident in certain cases (e.g., glioblastoma) and in some tumors treated with potent angiogenesis inhibitors, most tumors rely to a considerable extent on chronic angiogenesis to support their expansive growth. Still others may adapt to living in quasi-hypoxic environments where most cancer cells would perish.
Hallmark 6: activating invasion and metastasis
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


