Chapter 1 Biologic Basis of Radiation Oncology
What Is Radiation Biology?
Most of this chapter will be devoted to so-called classical radiobiology, that is, studies that largely predate the revolution in molecular biology of the 1980s and 1990s. Although the reader might be tempted to view this body of knowledge as rather primitive by today’s standards, relying too heavily on phenomenology, empiricism, and simplistic, descriptive models and theories, the real challenge is to integrate the new biology into the already-existing framework of classical radiobiology; this will be discussed in detail in Chapter 2.
Radiotherapy-Oriented Radiobiology: A Conceptual Framework
Therapeutic Ratio
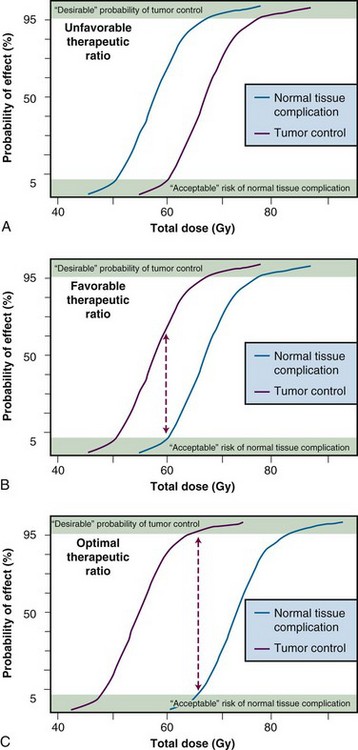
The concept of therapeutic ratio is best illustrated graphically, by making a direct comparison of dose-response curves for both tumor control and normal tissue complication rates plotted as a function of dose. Examples of this approach are shown in Figure 1-1, for cases in which the therapeutic ratio is either “unfavorable,” “favorable,” or “optimal,” bearing in mind that these are theoretical curves. Actual dose-response curves derived from experimental or clinical data are much more variable, particularly for tumors, which tend to show much shallower dose responses.1 This serves to underscore how difficult it can be in practice to assign a single numerical value to the therapeutic ratio in any given situation.
Radiation Biology “Continuum”
There is a surprising continuity between the physical events that occur in the first picosecond (or less) after ionizing radiation interacts with biologic material and the ultimate consequences of that interaction on tissues. The consequences themselves may not become apparent until days, weeks, months, or even years after the radiation exposure. Some of the important steps in this radiobiology “continuum” are listed in Table 1-1. The orderly progression from one stage of the continuum to the next—from physical to physicochemical to biochemical to biologic—is particularly noteworthy not only because of the vastly different time scales over which the critical events occur, but also because of the increasing biologic complexity associated with each of the endpoints or outcomes. Each stage of the continuum also offers a unique radiobiologic window of opportunity: the potential to intervene in the process and thereby modify all the events and outcomes that follow.

Tissue Heterogeneity
Normal tissues, being composed of more than one type of cell, are somewhat heterogeneous, and tumors, owing both to the genetic instability of individual tumor cells and to microenvironmental differences, are very heterogeneous. Different subpopulations of cells have been isolated from many types of human and experimental cancers, and these may differ in antigenicity, metastatic potential, sensitivity to radiation therapy and chemotherapy, and so on.2,3 This heterogeneity is manifest within a particular patient, and to a much greater extent, between patients with otherwise similar tumors.
What are the practical implications of normal tissue and tumor heterogeneity? First, if one assumes that normal tissues are the more uniform and predictable in behavior of the two, then tumor heterogeneity is responsible, either directly or indirectly, for most radiotherapy failures. If so, this suggests that a valid clinical strategy might be to identify the radioresistant subpopulation(s) of tumor cells and then tailor therapy specifically to cope with them. This approach is much easier said than done. Some prospective clinical studies now include one or more pretreatment determinations of, for example, the extent of tumor hypoxia4 or the potential doubling time of tumor clonogens5 as criteria for assigning patients to different treatment groups.
“Powers of Ten”
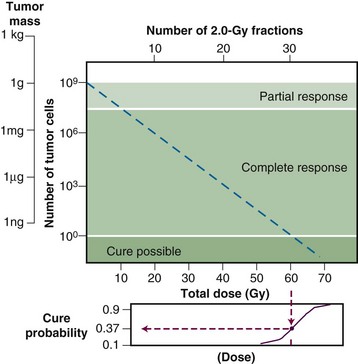
The tumor control probability for a given fraction of surviving cells is not particularly helpful if the total number of cells at risk is unknown, however, and this is where an understanding of logarithmic relationships and exponential cell killing is useful. Based on the resolution of existing tools and technology for cancer detection, let us assume that a 1-cm3 (1-g) tumor mass can be identified reliably. A tumor of this size has been estimated to contain approximately 109 cells,6 admittedly a theoretical value that assumes all cells are perfectly “packed” and uniformly sized and that the tumor contains no stroma. A further assumption, that all such cells are clonogenic (rarely, if ever, the case), suggests that at least 9 logs of cell killing would be necessary before any appreciable tumor control (about 37%) would be achieved, and 10 logs of cell killing would be required for a high degree of tumor control (i.e., 90%).
After the first log or two of cell killing, however, some tumors respond by shrinking, a partial response. After two to three logs of cell killing, the tumor may shrink to a size below the current limits of clinical detection, that is, a complete response. Although partial and complete responses are valid clinical endpoints, a complete response does not necessarily mean tumor cure. At least six more logs of cell killing would be required before any significant probability of cure would be expected. This explains why radiation therapy is not halted if the tumor “disappears” during the course of treatment; this concept is illustrated graphically in Figure 1-2.
Radiation Biology and Therapy: The First 50 Years
In less than 4 years after the discovery of x rays by Roentgen,7 radioactivity by Becquerel,8 and radium by the Curies,9 the new modality of cancer treatment known as radiation therapy claimed its first apparent cure of skin cancer.10 Today, more than a century later, radiotherapy is most commonly given as a series of small daily dose fractions of approximately 1.8 to 2 Gy each, 5 days per week, over a period of 5 to 7 weeks, to a total dose of 50 to 70 Gy. Although it is true that the historical development of this conventional radiotherapy schedule was empirically based, there were a number of early radiobiologic experiments that suggested this approach.
In the earliest days of radiotherapy, both x rays and radium were used for cancer treatment. Because of the greater availability and convenience of using x-ray tubes and the higher intensities of radiation output achievable, it was fairly easy to deliver large single doses in short overall treatment times. Thus, from about 1900 into the 1920s, this “massive dose technique”11 was a common way of administering radiation therapy. Unfortunately, normal tissue complications were quite severe. To make matters worse, the rate of local tumor recurrence was still unacceptably high.
As early as 1906, Bergonié and Tribondeau12 observed histologically that the immature dividing cells of the rat testis showed evidence of damage at lower radiation doses than the mature nondividing cells. Based on these observations, the two researchers put forth some basic “laws” stating that x rays were more effective on cells that were: (1) actively dividing, (2) likely to continue to divide indefinitely, and (3) poorly differentiated.12 Because tumors were already known to contain cells that not only were less differentiated but also exhibited greater mitotic activity, Bergonié and Tribondeau reasoned that several radiation exposures might preferentially kill these tumor cells but not their slowly proliferating, differentiated counterparts in the surrounding normal tissues.
The end of common usage of the single-dose technique in favor of fractionated treatment came during the 1920s as a consequence of the pioneering experiments of Claude Regaud and colleagues.13 Using the testis of the rabbit as a model tumor system (because the rapid and unlimited proliferation of spermatogenic cells simulated to some extent the pattern of cell proliferation in malignant tumors), Regaud14 showed that only through the use of multiple radiation exposures could animals be completely sterilized without producing severe injury to the scrotum. Regaud15 suggested that the superior results afforded the multifraction irradiation scheme were related to alternating periods of relative radioresistance and sensitivity in the rapidly proliferating germ cells. These principles were soon tested in the clinic by Henri Coutard,16,17 who used fractionated radiotherapy for the treatment of head and neck cancers, with spectacularly improved results. Mainly as a result of these and related experiments, fractionated treatment subsequently became the standard form of radiation therapy.
Time-dose equivalents for skin erythema published by Reisner,18 Quimby and MacComb,19 and others20,21 formed the basis for the calculation of equivalents for other tissue and tumor responses. By plotting the total doses required for each of these “equivalents” for a given level of effect in a particular tissue, as a function of a treatment parameter such as overall treatment time, number of fractions, dose per fraction, and so on, an isoeffect curve could be derived. All time-dose combinations that fell along such a curve would, theoretically, produce tissue responses of equal magnitude. Isoeffect curves, relating the total dose to the overall treatment time, derived in later years from some of these data,22 are shown in Figure 1-3.
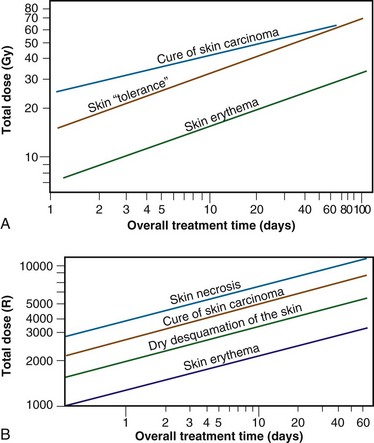
Figure 1-3 Isoeffect curves relating the log of the total dose to the log of the overall treatment time for various levels of skin reaction and the cure of skin cancer. A, Isoeffect curves were constructed by Cohen on the basis of a survey of earlier published data on radiotherapy equivalents.21–26 The slope of the curves for skin complications was 0.33, and the slope for tumor control was 0.22. B, The Strandqvist28 isoeffect curves were first published in 1944. All lines were drawn parallel and had a common slope of 0.33.
A, Adapted from Cohen L: Radiation response and recovery: Radiobiological principles and their relation to clinical practice. In Schwartz E, editor: The Biological Basis of Radiation Therapy, Philadelphia, 1966, JB Lippincott, p 208; B, adapted from Strandqvist M: Studien uber die kumulative Wirkung der Roentgenstrahlen bei Fraktionierung, Acta Radiol Suppl 55:1, 1944.
The first published isoeffect curves were produced by Strandqvist23 in 1944, and are also shown in Figure 1-3. When transformed on log-log coordinates, isoeffect curves for a variety of skin reactions, and the cure of skin cancer, were drawn as parallel lines, with common slopes of 0.33. These results implied that there would be no therapeutic advantage to using prolonged treatment times (i.e., multiple small fractions versus one or a few large doses) for the preferential eradication of tumors while simultaneously sparing normal tissues.24 It was somewhat ironic that the Strandqvist curves were so popular in the years that followed, when it was already known that the therapeutic ratio did increase (at least to a point) with prolonged, as opposed to very short, overall treatment times. However, the overarching advantage was that these isoeffect curves were quite reliable at predicting skin reactions, which were the dose-limiting factors at that time.
The “Golden Age” of Radiation Biology and Therapy: The Second 50 Years
Perhaps the defining event that ushered in the golden age of radiation biology was the publication of the first survival curve for mammalian cells exposed to graded doses of ionizing radiation.25 This first report of a quantitative measure of intrinsic radiosensitivity of a human cell line (HeLa, derived from a cervical carcinoma26) was published by Puck and Marcus25 in 1956. To put this seminal work in the proper perspective, however, it is first necessary to review the physicochemical basis for why ionizing radiation is toxic to biologic materials.
The Interaction of Ionizing Radiation with Biologic Materials
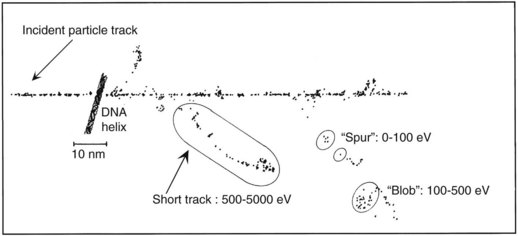
As mentioned in the introductory section of this chapter, ionizing radiation deposits energy as it traverses the absorbing medium through which it passes. The most important feature of the interaction of ionizing radiation with biologic materials is the random and discrete nature of the energy deposition. Energy is deposited in increasingly energetic packets referred to as “spurs” (100 eV or less deposited), “blobs” (100 to 500 eV), or “short tracks” (500 to 5000 eV), each of which can leave from approximately three to several dozen ionized atoms in its wake. This is illustrated in Figure 1-4, along with a segment of (interphase) chromatin shown to scale. The frequency distribution and density of the different types of energy deposition events along the track of the incident photon or particle are measures of the radiation’s linear energy transfer or LET (see also the “Relative Biologic Effectiveness” section, later). Because these energy deposition events are discrete, it follows that although the average amount of energy deposited in a macroscopic volume of biologic material may be rather modest, the distribution of this energy on a microscopic scale may be quite large. This explains why ionizing radiation is so efficient at producing biologic damage; the total amount of energy deposited in a 70-kg human that will result in a 50% probability of death is only about 70 calories, about as much energy as is absorbed by drinking one sip of hot coffee.27 The key difference is that the energy contained in the sip of coffee is uniformly distributed, not random and discrete.
Any and all cellular molecules are potential targets for the localized energy deposition events that occur in spurs, blobs, or short tracks. Whether the ionization of a particular biomolecule results in a measurable biologic effect depends on a number of factors, including how probable a target the molecule represents from the point of view of the ionizing particle, how important the molecule is to the continued health of the cell, how many copies of the molecule are normally present in the cell and to what extent the cell can react to the loss of “working copies,” and how important the cell is to the structure or function of its corresponding tissue or organ. DNA, for example, is obviously an important cellular macromolecule and one that is present only as a single double-stranded copy. On the other hand, other molecules in the cell may be less crucial to survival yet are much more abundant than DNA, and therefore have a much higher probability of being hit and ionized. The most abundant molecule in the cell by far is water, comprising some 80% to 90% of the cell on a per weight basis. The highly reactive free radicals formed by the radiolysis of water are capable of adding to the DNA damage resulting from direct energy absorption by migrating to the DNA and damaging it indirectly. This mechanism is referred to as “indirect radiation action” to distinguish it from the aforementioned “direct radiation action.”28 The direct and indirect action pathways for ionizing radiation are illustrated below.
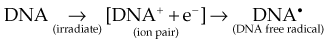
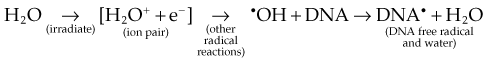
The most highly reactive and damaging species produced by the radiolysis of water is the hydroxyl radical (•OH), although other free radical species are also produced in varying yields.29,30 Ultimately, it has been determined that cell killing by indirect action constitutes some 70% of the total damage produced in DNA for low LET radiation.
The •OH radical is capable of both abstraction of hydrogen atoms from other molecules and addition across carbon-carbon or other double bonds. More complex macromolecules that have been converted to free radicals can undergo a series of transmutations in an attempt to rid themselves of unpaired electrons, many of which result in the breakage of nearby chemical bonds. In the case of DNA, these broken bonds may result in the loss of a base or an entire nucleotide or in frank scission of the sugar phosphate backbone, involving one or both DNA strands. In some cases, chemical bonds are first broken but then rearranged, exchanged, or rejoined in inappropriate ways. Bases in DNA may be modified by the addition of one or more hydroxyl groups (e.g., the base thymine converted to thymine glycol), pyrimidines may become dimerized, and/or the DNA may become cross-linked to itself or to associated protein components. And again, because the initial energy deposition events are discrete, the free radicals produced also are clustered and therefore undergo their multiple chemical reactions and produce multiple damages in a highly localized area. This has been termed the “multiply-damaged site”31 or “cluster”32 hypothesis. Examples of the types of damage found in irradiated DNA are shown in Figure 1-5.
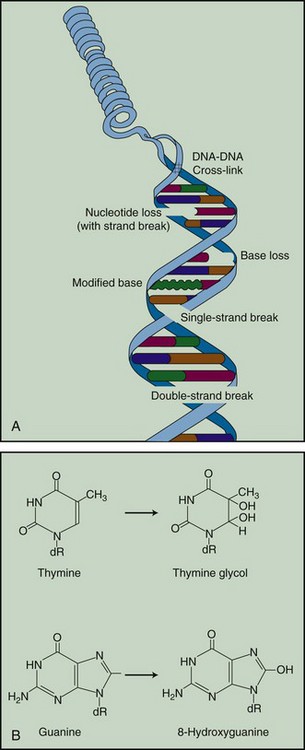
Biochemical Repair of DNA Damage
The study of DNA repair in mammalian cells received a significant boost during the late 1960s with publications by Cleaver33,34 that identified the molecular defect responsible for the human disease xeroderma pigmentosum (XP). Patients with XP are exquisitely sensitive to sunlight and highly cancer-prone (particularly to skin cancers). Cleaver showed that cells derived from such patients were likewise sensitive to UV radiation and defective in the nucleotide excision repair pathway (discussed later). These cells were not especially sensitive to ionizing radiation, however. Several years later, Taylor and colleagues35 reported that cells derived from patients with a second cancer-prone disorder called ataxia telangiectasia (AT) were extremely sensitive to ionizing radiation and radiation-mimetic drugs but not to UV radiation. In the years that followed, cell cultures derived from patients with these two conditions were used to help elucidate the complicated processes of DNA repair in mammalian cells.
Today, many rodent and human genes involved in DNA repair have been cloned.36 Nearly 20 distinct gene products participate in excision repair of base damage, and even more are involved in the repair of strand breaks. Many of these proteins function as component parts of larger repair complexes; some of these parts are interchangeable and participate in other DNA repair and replication pathways as well. It is also noteworthy that some are not involved with the repair process per se but rather link DNA repair to other cellular functions, including transcription, cell cycle arrest, chromatin remodeling, and apoptosis.37 This attests to the fact that the maintenance of genomic integrity results from a complex interplay between not only the repair proteins themselves but between others that serve as damage sensors, signaling mediators and transducers, and effectors.
For example, the defect responsible for the disease AT is not in a gene that codes for a repair protein but rather in a gene that participates in damage recognition and in a related pathway that normally prevents cells from entering S phase of the cell cycle and beginning DNA synthesis while residual DNA damage is present. This was termed the G1/S checkpoint response.38 Because of this genetic defect, AT cells do not experience the normal G1 arrest after irradiation and enter S phase with residual DNA damage. This accounts both for the exquisite radiosensitivity of AT cells and the resulting genomic instability that can lead to carcinogenesis.
What is known about the various types of DNA repair in mammalian cells is outlined below.
Base Excision Repair
The repair of base damage is initiated by DNA repair enzymes called DNA glycosylases, which recognize specific types of damaged bases and excise them without otherwise disturbing the DNA strand.39 The action of the glycosylase itself results in the formation of another type of damage observed in irradiated DNA—an apurinic or apyrimidinic (AP) site. The AP site is then recognized by another repair enzyme, an AP endonuclease, which nicks the DNA adjacent to the lesion. The resulting strand break becomes the substrate for an exonuclease, which removes the abasic site, along with a few additional bases. The small gap that results is patched by DNA polymerase, which uses the opposite, hopefully undamaged, DNA strand as a template. Finally, DNA ligase seals the patch in place.
Nucleotide Excision Repair
The DNA glycosylases that begin the process of base excision repair do not recognize all known forms of base damage, however, particularly bulky or complex lesions.39 In such cases, another group of enzymes, termed structure-specific endonucleases, initiate the excision repair process. These repair proteins do not recognize the specific lesion but are thought instead to recognize more generalized structural distortions in DNA that necessarily accompany a complex base lesion. The structure-specific endonucleases incise the affected DNA strand on both sides of the lesion, releasing an oligonucleotide fragment made up of the damage site and several bases on either side of it. Because a longer segment of excised DNA—including both bases and the sugar phosphate backbone—is generated, this type of excision repair is referred to as nucleotide excision repair to distinguish it from base excision repair (described earlier), where the initial step in the repair process is removal of the damaged base only. After this step, the remainder of the nucleotide excision repair process is similar to that of base excision repair; the gap is then filled in by DNA polymerase and sealed by DNA ligase. Overall, however, nucleotide excision repair is a much slower process, with a half-time of approximately 12 hours.
For both types of excision repair, active genes in the process of transcription are repaired preferentially and more quickly. This has been termed transcription-coupled repair.40
Strand Break Repair
Despite the fact that unrepaired or mis-rejoined strand breaks, particularly DSBs, often have the most catastrophic consequences for the cell in terms of loss of reproductive integrity,41 the way in which mammalian cells repair strand breaks has been more difficult to elucidate than the way in which they repair base damage. Much of what was originally discovered about these repair processes is derived from studies of x ray–sensitive rodent cells and microorganisms that were subsequently discovered to harbor specific defects in strand break repair. Since then, other human genetic diseases characterized by DNA repair defects have been identified and are also used to help probe these fundamental processes.
A genetic technique known as complementation analysis allows further characterization of genes involved in DNA repair. Complementation analysis involves the fusion of different strains of cells possessing the same phenotypic defect and subsequent testing of the hybrid cell for the presence or absence of this phenotype. Should two cell types bearing the defect yield a phenotypically normal hybrid cell, this implies that the defect has been “complemented” and that the defective phenotype must result from mutations in at least two different genes. With respect to DNA strand break repair, eight different genetic complementation groups have been identified in rodent cell mutants. For example, a mutant Chinese hamster cell line, designated EM9, seems especially radiosensitive owing to delayed repair of DNA single-strand breaks, a process that is normally complete within minutes of irradiation.42
With respect to the repair of DNA DSBs, the situation is more complicated in that the damage on each strand of DNA may be different, and, therefore, no intact template would be available to guide the repair process. Under these circumstances, cells depend on genetic recombination (nonhomologous end joining or homologous recombination43) to cope with the damage.
The BRCA1 and BRCA2 gene products are also implicated in HR (and, possibly, NHEJ as well) because they interact with the Rad51 protein. Defects in these genes are associated with hereditary breast and ovarian cancer.44
Mismatch Repair
The primary role of mismatch repair (MMR) is to eliminate from newly synthesized DNA errors such as base/base mismatches and insertion/deletion loops caused by DNA polymerase.45 Descriptively, this process consists of three steps: mismatch recognition and assembly of the repair complex, degradation of the error-containing strand, and repair synthesis. In humans, MMR involves at least five proteins, including hMSH2, hMSH3, hMSH6, hMLH1, and hPMS2, as well as other members of the DNA repair and replication machinery.
One manifestation of a defect in mismatch repair is microsatellite instability, mutations observed in DNA segments containing repeated sequence motifs.46 Collectively, this causes the cell to assume a hypermutable state (“mutator phenotype”) that has been associated with certain cancer predisposition syndromes, in particular, hereditary nonpolyposis colon cancer (HNPCC, sometimes called Lynch syndrome).47,48
Cytogenetic Effects of Ionizing Radiation
Most chromosome aberrations result from an interaction between two (or more; discussed later) damage sites, and, therefore, can be grouped into three different types of “exchange” categories. A fourth category is reserved for chromosome aberrations thought to result from a single damage site.49 These categories are described below, and representative types of aberrations from each category are shown in Figure 1-6:
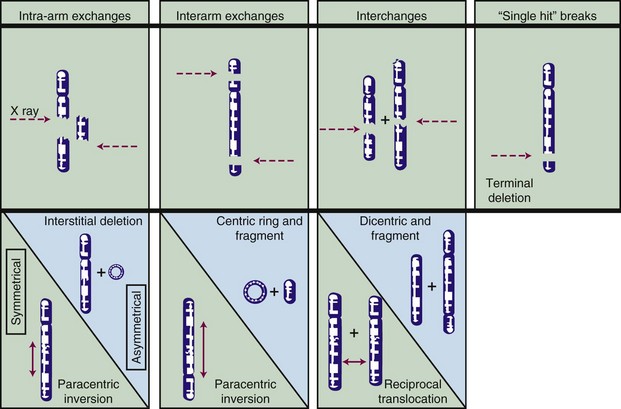
These four categories can be further subdivided according to whether the initial radiation damage occurred before or after the DNA is replicated during S phase of the cell cycle (a chromosome-type vs. a chromatid-type of aberration, respectively) and, for the three exchange categories, whether the lesion interaction was symmetric or asymmetric. Asymmetric exchanges always lead to the formation of acentric fragments that are usually lost in subsequent cell divisions and therefore are nearly always fatal to the cell. These fragments may be retained transiently in the cell’s progeny as extranuclear chromatin bodies called micronuclei. Symmetric exchanges are more insidious in that they do not lead to the formation of acentric fragments and the accompanying loss of genetic material at the next cell division, they are sometimes difficult to detect cytologically, and they are not invariably lethal to the cell. As such, they will be transmitted to all the progeny of the original cell. Some types of symmetric exchanges (a reciprocal translocation, for example) have been implicated in radiation carcinogenesis, insofar as they have the net effect of either bringing new combinations of genes together or of separating preexisting groups of genes.27 Depending on where in the genome the translocation takes place, genes normally on could be turned off, or vice versa, possibly with adverse consequences.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


