In the BM, hematopoietic stem cells (HSCs) develop in a complex microenvironment of stromal cells (fibroblasts, smooth muscle cells, endothelial cells, adipocytes, osteoblasts, osteoclast, & MΦ) & extracellular matrix
BM comprises hematopoietic tissue islands & adipose cells surrounded by vascular sinuses
In nl adults, the ratio of fat cells to hematopoietic elements is 1:1
Thin-walled sinusoids are lined by endothelial cells, & differentiated blood cells enter the circulation by transcellular migration through endothelial cells
HSCs: 2 cardinal features (1) pluripotency (can give rise to all mature blood cell types) & (2) self-renewal capacity
HSCs give rise to all formed elements of blood – RBCs, granulocytes, monocytes, plt, & lymphocytes
HSCs give rise to two kinds of multipotent cells, the common lymphocyte progenitors (CLPs) & common myeloid progenitors (CMPs)
CMPs give rise to committed progenitors called colony forming units (CFUs) that develop into specific mature cells
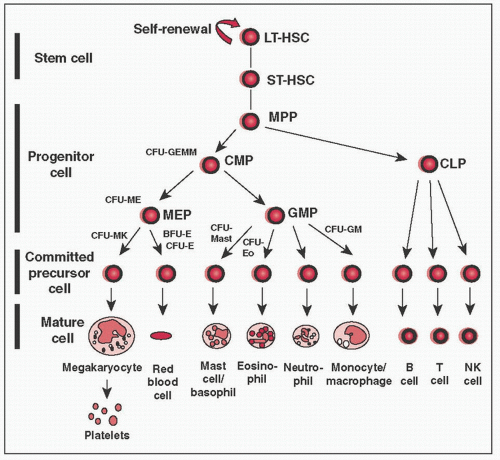
Figure 28-1 Cantor, A. B. et al. ASH-SAP 2010;2010:331-372.
CLP = common lymphoid progenitor, CFU = colony forming unit, CMP = common myeloid progenitor, GMP = granulocyte-MΦ progenitor, LT-HSC = long-term repopulating hematopoietic stem cell, MEP = megakaryocyte-erythroid progenitor, MPP = multipotential progenitor cell, NK = natural killer, ST-HSC = short-term repopulating hematopoietic stem cell
Various factors modulate BM response to physiologic needs & production of mature blood cells
Cytokines (IL-1, IL-2, IL-3, IL-5, IL-6, & IL-7) & chemokines
Sites: Preferred site is the posterior iliac spine. Alternate sites include anterior iliac crest or sternum.
BM aspiration: Advance needle through cortex into medullary space. Aspirate marrow using syringe & spread on glass slides. Additional cell suspensions used for cytogenetics (karyotype & FISH), flow cytometry, & molecular studies (eg, FLT-3 or JAK-2 Mt). Use aspirate to assess morphology of hematopoietic cells.
BM bx: Use longer needle (such as Jamshidi needle) to cut a cylinder of bone from the medullary space. W/bx, assess BM architecture, cellularity, iron stores, cell lineage.
Risks: Hemorrhage, infxn
Cellularity: 100 – pt age = nl % cellularity.
Myeloid: Erythroid ratio: Nl range 1.2:1-5:1 → assess on bx or aspirate. >5:1 ratio – physiologic stress (infxn), <1.2:1 ratio – erythroid response to anemia (hemolysis).
Megakaryocytes: Assess number & appearance. Nl cells are large, multilobulated, irregularly lobed nuclei.
Myeloid maturation (see below), erythroid maturation (see below). Excess blasts or differentiation arrest suggestive of leukemia
Dysplasia: Nuclear & cytoplasmic blebs & dysmorphic nuclei.
Erythroid dysplasia: Multinucleated erythroid precursors, nuclear-cytoplasmic dyssynchrony, RS
Myeloid dysplasia: Hypersegmented neutrophils, hyposegmented neutrophils (Pseudo-Pelger-Huet), hypogranular neutrophils, Auer rods, dimorphic granules (basophilic & eosinophilic granules in eosinophils)
Megakaryocyte dysplasia: Hyper-/hypolobulated megakaryocytes, micromega-karyocytes (smaller in size)
Lymphocytes & plasma cells: Benign lymphoid population, more T cells than B cells. Abnl, such as a monotonous B-cell lymphocyte population, is suggestive of lymphoma. Plasma cells <2% of BM cells in nl adult, ↑ % in plasma cell dyscrasia
Iron stores: Can be ↑ (hemochromatosis) or ↓ (IDA)
Other cells: Marrow infiltration by tumor or fibrosis (“myelophthisis”)
Erythroid maturation: The nucleus becomes progressively smaller and nuclear chromatin more condensed. The cytoplasm gradually loses the bluish color from RNA, w/c is replaced by the pink-staining Hb.
Stages of maturation:
Proerythroblast, basophilic erythroblast, polychromatophilic erythroblast, orthochromatic erythroblast (normoblast), polychromatic erythrocyte (reticulocyte), erythrocyte
Myeloid maturation: Granulocytes are the precursors & mature forms of leukocytes w/neutrophilic, eosinophilic, or basophilic granules in their cytoplasm in mature forms
Stages of maturation:
Myeloblast, promyelocyte, myelocyte, metamyelocyte, band cell, mature cell (basophil, neutrophil, eosinophil)
|
S/s: General anemia sxs. Pica (uncommon), stomatitis, glossitis, koilonychia, esophageal webs: Plummer-Vinson Syndrome (rarer).
Lab W/u: ↓↓ Ferritin (< 15 µg/L diagnostic; >100 µg/L unlikely).High nl to ↑ TIBC. ↓↓ Fe sat (<10%). Nl MCV (early) → ↓ to ↓↓ MCV (adv). ↑ RDW. Experimental: ↓ Hepcidin. ↑ Soluble transferrin receptor. Anemia/Fe parameters improve w/Fe administ.
Tx: Uncomplicated/Mild Fe Deficiency: PO Fe. Ferrous salts ↑ solubility & absorption but ↑ GI tox. Ascorbic acid → ↑ absorption. ↓ Absorption w/antacids, full meals, whole grains, tannins & Ca supplements. Fe sulfate = 66 mg elemental Fe per 325-mg tab.
Complicated Fe Deficiency: Parenteral Fe in true ↓ absorption, gastric or duodenal resection, ↑ Fe deficit, PO Fe intolerance & chronic bleeding.
Fe Dextran: ↓↓cost. Can replete Fe in one infusion. ↑ Risk anaphylaxis (11.3/1000000) → small test dose. Fe Sucrose & Fe Gluconate: ↓↓↓ Rate anaphylaxis → no test dose. A/w arthralgias/myalgias. ↑↑ cost.
Hypoproliferative anemia (functional Fe def) a/w chronic inflammation: CTD, other autoimmune disorders, chronic infxn, & malignancy.
Also in post-trauma/-surgery & critical illness. Commonest anemia in hospital.
Pathogenesis: ↑ Cytokines (TNFα, IL-1, IL-6, IFNs) →↓ response to EPO in RBC precursor, ↓ EPO c/w anemia degree, & mod. ↓ RBC survival.
Altered Fe metabolism (Blood 2003;102:783): ↑ IL-6 →↑ hepcidin →↓ intestinal Fe absorption/↓ Fe release from enterocytes →↓ plasma Fe/Fe def.
Dx: Nl chromic. Nl/↓ MCV. Nl to ↑ RDW. ↓ to nl Fe. ↓ to low nl TIBC. ↓ Fe sat. Nl to ↑↑ ferritin. Experimental: Nl sol transferrin receptor/↑ Hepcidin.
Folate Deficiency: Duodenum/proximal jejunum absorption. No A/w neuro sxs.
Etiologies: ↓ Intake, ↓ absorption, ↑ use (chronic hemolysis), drug interaction. ↑Risk in alcoholics, elderly, TPN w/o supplemental folate, duodenal or jejunal resection/infiltration, celiac disease, chronic hemolysis.
Dx: ↓ Serum folate ± RBC folate (may better reflect folate def but also ↓ in B12 Def), ↑ homocysteine, nl methylmalonic acid (MMA). Also a/w falsely ↓ B12 in some cases.
B12 Deficiency: Multiple steps for dietary B12 absorption in terminal ileum.
↓Intake (no animal products/vegans). Hypochlorydia (age, PPIs, atrophic gastritis). ↓ Pancreatic Proteases. ↓ Binding B12 & IF (bacterial overgrowth, Diphyllobothrium, pernicious anemia, & gastric resection). ↓ Absorption w/Meds (cholestyramine, colchicine, & metformin). Ileal Resection/Dysfunction (Celiac, NHL, Crohn’s, etc.).
A/w neuropsych sxs: ↓ vibriosensation, spastic paralysis, psychosis/dementia.
Full B12 stores take longer to deplete than folate stores: ˜ few y.
Clinical B12 Def: ↓ B12, ↑↑ homocysteine, ↑↑ MMA.
Tx: Beware folate supp only correcting anemia & not neuro sxs. IM B12 1 mg/d × 14 d → 1 mg/wk until anemia resolves → 1 mg/mo.
Other Megaloblastic Anemias: Pyrimidine Analog
Macrocytic Anemias w/Similar Morphologic Δ’s: Myelodysplasia, erythroleukemia, Lesch-Nyhan Syndrome, Hereditary Orotic Aciduria.
Echinocytes in ↑↑ CKD. BM normocellular (hypocellular in long-standing 2° ↑ PTH → osteitis fibrosa). Tx: Follow Fe Sat/ferritin & replete Fe stores. ESA Tx: Goal Hb recs Δ’d in recent y. Balance ↑ QoL vs. ↑ mort, CV, thrombotic outcomes? → esp w/solid tumor. TREAT (N Engl J Med 2009;361:2019). CHOIR (N Engl J Med 2006;355:2085). EPO-CAN-20 (J Clin Oncol 2007;25:1027).
EPO Resistance: ˜25% HD pts w/some EPO resistance. A/w ↑ inflamm (↑ IL-6).
RBC intrinsic vs. extrinsic disorder. Intravascular vs. extravascular (most hemolysis in-between on spectrum & manifest s/s of both.
Inherited vs. acquired. Compensated vs. decompensated.
Signs: Gen- pale, jaundice, ↓ Hb (decompensated), ↑ indirect bili., ↑ LDH, ↑ retic (RI > 2 & ↑ absolute number if nl BM response), ↓ haptoglobin. ↑ urobilinogen.
Extravascular- splenomegaly, pigmented cholelithiasis.
|
G6PD A: 10-15% African-American males. Unstable enzyme → ↓ activity in aged RBCs, not retics. Vital oxidants may be continued b/c ↑↑ retics.
G6PD Mediterranean: B variant. Both retics/mature RBCs affected → ↑↑↑ sev. course. Must stop oxidant agent/process.
Other Variants: ↓ G6PD activity + marked instability/↓ production → mature RBC & retics affected. ↑↑ severity. Seen in Asia/Mediterranean area.
Tx depends on severity. Consider folate supplementation & splenectomy.
Hereditary spherocytosis (HS) ↑ in N. Europeans: ˜1/2000. Variable penetrance. 75% autosomal dominant. Etiologies: 30-45% ↓ ankyrin & spectrin, 30% ↓ spectrin, 20% band 3 Mt. ↓ Protein 4.2 as well.
AbNl protein → unstable membrane → ↓ bilayer/↓surface area → spherocyte.
Range: Asymptomatic → Symptomatic sev. A/w aplastic/megaloblastoid/hyperhemolytic crises.
Dx: Spherocytes, DAT-, ↑ MCHC, + osmotic fragility test, EMA-binding test.
Tx: Consider folic acid in ↑ hemolysis. Definitive Tx: Splenectomy if ↑↑ severity.
Defect spectrin dimerization/spectrin-protein 4.1 interaction → weakened cytoskeleton → deformation under shear stress → ↓ membrane fidelity.
Acanthocytosis: Liver disease → nonesterified cholesterol in RBC membrane → AbNl shape → splenic remodeling. A/w Zieve syndrome.
Abetalipoproteinemia: AbNl RBC membrane lipid structure.
Stomatocytosis: Inherited: AbNl RBC cation permeability. Acquired: CA, Alcoholism, & hepatobiliary disease. ↑ Thrombosis postsplenectomy.
Rh Deficiency/Null: No or ↓↓↓ Rh (RhCE, RhD, & Rh50). Autosomal recessive → Δ cation xport/RBC dehydration → spherocytes/stomatocytes.
Warm AIHA: 80-90% adult AIHA cases. IgG bind RBCs optimally at 37°C. ± C′ fixation. No agglutination. IgG-coated RBCs → removed by splenic mφ w/Fc receptors. 2° Warm AIHA a/w: HL, NHL, CTD (SLE), solid tumor (ovary), chronic inflammation, (UC) & drugs (α-methyldopa).
Tx: 1°: 1st-line steroids. 2nd-line splenectomy/rituximab. 3rd-line cyclophosphamide, AZA, MMF, CsA, danazol, alemtuzumab. ↓↓ Efficacy w/IVIg. 2°: SLE: 1st-line steroids. 2nd-line rituximab. ↓↓ Efficacy w/splenectomy. CLL: Steroids, R-CVP, R-CD, chlorambucil, rituximab, CsA, splenectomy, alemtuzumab. NHL: Tx malignancy. splenectomy in splenic MZL. Drugs: Stop agent; steroids.
Cold AutoAb: Mostly IgM optimally binding RBCs <37°C. A/w agglutination.
RBC destruction mediated by C′ C3b fixation → removal by splenic mφ/hepatic Kupffer cells. + Intravascular lysis w/terminal C′.
Cold Agglutinin: A/w B-cell LPD (monoclonal IgM against Carbohydrate I or i Ags). Cold agglutinin w/2° Cold AIHA (IgM): Mycoplasma & EBV.
Cold (Hemolysin) AIHA: 1°: Paroxysmal cold hemoglobinuria (IgG). 2°: Donath-Landsteiner Hemolytic Anemia (1/3 pediatric AIHA; a/w viral infxn; anti-P system IgG). Congenital/3° syphilis cold AIHA (IgG).
Tx: General-avoid cold; rituximab. No efficacy w/steroids/IVIg. Plasmapheresis acute ↓ IgM. Tx LPD. Donath-Landsteiner: Nl self-limited. ?Eculizumab; ?bortezomib.
DAT: IgG: Warm AIHA, hapten-mediated/drug adsorption or true autoAb. C3d: Warm AIHA w/↓IgG bound, cold agglutinin, paroxysmal cold hemoglobinuria or ternary/immune complex. IgG + C3d: Warm AIHA or true autoAb.
Xfusion is complex → autoAbs mask alloAbs (potentially sev.) → work w/blood bank to find compatible units. Obtain Hx of prior pregnancy/abortion/xfusion.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


