Introduction
The autoimmune polyglandular syndromes (APS) are uncommon constellations of organ-specific autoimmune diseases characterized by the occurrence of more than one autoimmune disease in an affected individual ( Table 22.1 ). Although autoimmune endocrine disorders commonly affect single organs, multiorgan autoimmune involvement of both endocrine and nonendocrine organs and tissues, secondary to loss of self-tolerance, is a characteristic feature of APS.
| APS I | APS II | |
|---|---|---|
| Comparative frequency Onset Heredity Gender Genetics Hypoparathyroidism Mucocutaneous candidiasis Ectodermal dysplasia Addison disease Type 1 diabetes Autoimmune thyroid disease Pernicious anemia Gonadal failure Females Males Vitiligo Alopecia Autoimmune hepatitis Malabsorption | Less common Infancy/early childhood Autosomal recessive Males = females AIRE gene; no HLA association 77%–89% 73%–100% 77% 60%–86% 4%–18% 8%–40% 12%–15% 30%–60% 7%–17% 4%–13% 27% 10%–15% 10%–18% | More common Late childhood, adulthood Polygenic Female predominance HLA associated; DR/DQ < 5% None None 70%–100% 41%–52% 70% 2%–25% 3.5%–10% 5% 4%–5% 2% Rare Rare |
Tolerance is a physiological state in which one’s immune system recognizes self-antigens and does not mount an immune response to self-antigens. If tolerance is not established or is lost, autoimmunity and subsequent disease may result. Although breakdown in self-tolerance remains mostly unexplained, improved understanding of the complex interplay between genetics and the environment, and the resultant aberrant immunological processes has identified a number of possible mechanisms. To comprehend these mechanisms, a brief overview of how self-tolerance is established and maintained is essential.
Mechanisms underlying tolerance
Introduction
The body’s first line of immune defense, after physiological barriers against invading foreign pathogens, is the innate arm of the immune system. Innate immunity is a nonspecific response mediated by expression of germ line genes, which does not require prior exposure to an antigen and provides immediate defense. However, the innate immune system does not exhibit immunologic memory and does not provide long-lasting protection. The next line of defense against foreign antigens is the adaptive immune system. Although the adaptive immune system assumes all exogenous antigens are potentially harmful, it produces antigen-specific responses. In a normal adaptive immune response; the host organism must differentiate self- from nonself-antigens; mount an immune response; eliminate or remove the inciting antigen; and protect the host from injury, organ dysfunction, and even death. Discrimination of self/nonself is carried out by the adaptive (specific) immune system by a mechanism that uses specific T-and B-cell surface receptors. T-cell and B-cell receptors recognize distinctive antigen peptides and epitopes, respectively, and are the keys to the specificity of the adaptive immune response. Whereas B cells and their receptors recognize soluble antigens on cell surfaces of pathogens, T cells and their receptors (TCRs) perceive short polypeptides only when presented by antigen presenting cells (APCs), using specialized cell-surface molecules encoded by the major histocompatibility complex (MHC). The human MHC is termed the human leukocyte antigen (HLA) complex. Class I MHC (e.g., HLA-A, HLA-B, and HLA-C molecules) is found on all nucleated cells, and presents endogenous peptides derived from the cytoplasm (such as from a pathogen infected cell) to CD8 T cells. Class II MHC (e.g., HLA-DP, HLA-DQ, and HLA-DR) molecules, on the other hand, present foreign antigens (peptides) that have been endocytosed and processed by APCs (such as B cells, dendritic cells, and macrophages) to CD4 T cells.
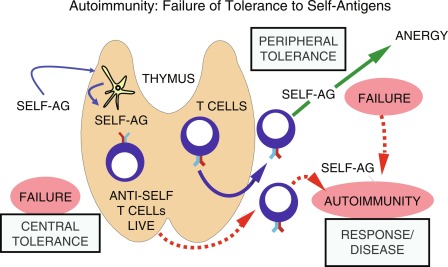
T cells are initially classified based on their cluster of differentiation (CD) surface proteins that bind differentially to antigens presented on class I (binds to CD8) and class II (binds to CD4) MHC. For example, CD8 T cells, also called cytotoxic T cells when activated, typically recognize antigens presented via class I MHC, and mediate an immune response specific to that antigen. Conversely, CD4 T cells, serving as helper or regulatory T cells, are activated when presented with peptides via class II MHC. Regulation of T-cell self-tolerance occurs at two distinct but interdependent levels: central tolerance and peripheral tolerance (described later) ( Fig. 22.1 ). Central tolerance occurs in the thymus, whereas peripheral tolerance occurs in both lymphoid and nonlymphoid tissues. Although many of the mechanisms involved in self-tolerance remain poorly understood, almost 2 decades of characterizing the autoimmune regulatory gene (AIRE) has improved our understanding of the pathways in the establishment and maintenance of self-tolerance. The AIRE gene encodes a transcription factor in the thymic medulla, which plays a critical role in establishing central tolerance (described in detail in the later part of this chapter). Deletions in the mouse AIRE homologue result in multiorgan autoimmunity, whereas mutations in the human AIRE gene result in APS I.
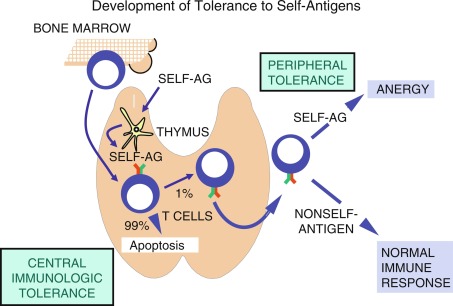
Tolerance is initially developed in utero with T and B cells playing important roles. T and B cells are produced continuously throughout life by hematopoietic stem cells of the bone marrow, with the T-cell precursors migrating to the thymus for further maturation. Although the thymus atrophies after puberty, residual thymic tissue may provide for T cell development throughout life.
Although T cells require exposure to small doses of antigen to achieve tolerance during their thymic development, larger doses of antigen are required to induce B-cell tolerance, and B-cell tolerance is often short lived. Tolerance is immunologically specific and induced in developing lymphocytes early in life; however, it can also be induced in mature lymphocytes when costimulatory signals are absent at the time of peptide interaction with TCR.
Central T-Cell Tolerance and AIRE
T cells are primarily educated to distinguish self and nonself when they develop in the thymus. In the thymic cortex, CD4 + CD8 + (double-positive) T cells bearing α/β TCRs that are able to bind to self-peptide/MHC complexes are selected to survive, whereas T cells whose TCRs fail to bind undergo apoptosis. As many as 99% of developing thymocytes undergo apoptosis and never reach the periphery (see Fig. 22.1 ). This is referred to as positive selection and is carried out by antigen-presenting cortical thymic nurse epithelial cells (cTECs), bearing MHC I and MHC II. T cells that bind to MHC I commit the developing T cell toward a CD8 T-cell pathway, whereas those that bind to MHC II develop into CD4 T cells. Positively selected cells migrate through the corticomedullary junction into the medulla where a secondary checkpoint occurs. Although T cells must be able to bind to MHC/self-peptide to establish tolerance, cells that bind too tightly to self-antigens are capable of inducing autoreactivity and therefore undergo negative selection and apoptosis. This process of positive selection of T cells for positive selection of MHC binding T cells (in the thymic cortex) and negative selection of T cells tightly bound to self-antigens (in the medulla), accounts for central (thymic) immunological tolerance. The single positive T cells expressing either CD4 or CD8 then migrate to the periphery and secondary lymphoid organs.
Medullary thymic epithelial cells (mTECs) are specifically involved in the process of negative selection of T cells and express an enormously diverse range of peripheral tissue–specific antigens (TSAs) for presentation to the developing T cells. This has been referred to as promiscuous gene expression (PGE) and is mediated by AIRE, a transcriptional regulator. Notably, AIRE is expressed in a small population of mature mTECs, with high levels of CD80 and MHC II. Unlike traditional transcriptional regulators, it does not bind to deoxyribonucleic acid (DNA) segments but activates ribonucleic acid (RNA) polymerases, and elongates TSA RNA transcripts. This process is also mediated by interaction of AIRE with other transcriptional regulators. Another transcriptional regulator that is involved in negative T-cell selection in the thymus is the FEZ family zinc finger protein 2 (FEZF2), which is involved in AIRE-independent TSA expression in mTECs. Studies have shown that AIRE and FEZF2 share complementary and parallel function in establishing and maintaining central tolerance. In addition to regulation of TSA expression in mTECs, AIRE is involved in thymic selection and differentiation of autoreactive CD4 T cells into regulatory T cells (Tregs), and upregulation of chemokines that aid in thymocyte migration. Extrathymic expression of AIRE in tissues, such as the bone marrow, has also been shown to aid in peripheral tolerance by inducing anergy of CD4 and CD8 T cells.
Peripheral T-Cell Tolerance
Once naïve T cells enter the circulation or secondary lymphoid organs (e.g., lymph nodes and spleen) and recognize specific antigens, they require additional cosignals to become activated. The first signal involves the interaction of antigen-peptides bound to the MHC molecules on the surface of APCs with TCRs on the surface of CD4 and CD8 T-cells. CD4 and CD8 molecules on these T-cell subsets serve as antigen-nonspecific coreceptors binding to nonpolymorphic portions of the class II and class I MHC molecules, respectively. The second signal is antigen nonspecific and is provided by the B7.1 (CD80) and B7.2 (CD86) molecules of the APC interacting with the CD28 molecule on the T-cell surface ( Fig. 22.3 ). In addition to CD80/CD86/CD28 interactions, there are other costimulatory signals that have important roles in T-cell development.
When T cells perceive both signals (MHC-antigen with TCR and B7-CD28), a cascade of intracellular signaling events occur, leading to T-cell activation. Activated cytotoxic CD8 T cells mediate direct lysis of target cells, whereas activated CD4 T cells lead to expression of numerous cytokines, cytokine receptors, and cytotoxic T-lymphocyte antigen 4 (CTLA-4). CTLA-4 is homologous to CD28 and competes with CD28 for binding to B7.1/B7.2. CTLA-4 expression by the activated T cell and its interaction with B7.1/B7.2 provide an immunosuppressive/immunoregulatory signal to the T-cell, thereby downregulating the T-cell responses. Thus CTLA-4 and CD28 though homologous, act antithetically: B7.1/B7.2-CD28 turns on T cells, whereas B7.1/B7.2-CTLA-4 downregulates the T cell (see Fig. 22.2 ).
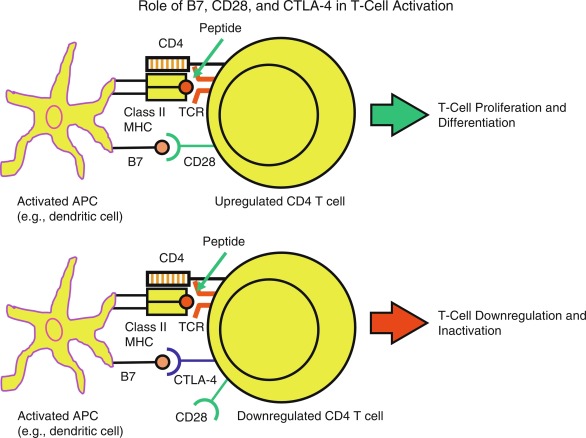
The requirement for two signals to activate naïve T cells mainly accounts for peripheral T-cell tolerance. When the naïve T cell perceives antigen peptide presented by MHC molecules without the necessary costimulatory signal (e.g., B7.1/B7.2-CD28), the T cell becomes unresponsive. This state of unresponsiveness is termed anergy ; anergic T cells are generally not restimulated with antigen peptide displayed by the APCs. T cells may also undergo apoptosis (programmed cell death) to be removed completely from the T-cell repertoire. Tolerance may also exist because the TCR does not encounter the relevant peptide, and this has been termed T-cell ignorance .
Another mechanism whereby T-cell tolerance is mediated is the interaction of programmed death 1 (PD-1) receptor on T cells and its ligands PD-L1 and PD-L2. These interactions bring about inhibition of T-cell effector functions in an antigen-specific manner. PD-1 signaling can also mediate the conversion of naïve T cells to Tregs.
Helper CD4 T cells are classically divided into two distinct lineages: (1) Th1 cells, which activate cell-mediated and some antibody responses, and (2) Th2 cells, which predominantly activate antibody-mediated responses. However, additional T-cell lineages exist (i.e., Th17 cells, T-follicular helper cells, and regulatory T cells) and recent data have described remarkable plasticity in their cytokine expression, suggesting shifting T-cell functionalities, depending on environmental cues. Although overtly simplistic, Th1 subsets secrete predominantly proinflammatory cytokines, such as interleukin (IL)-2, interferon-gamma (IFN-γ), tumor necrosis factor-beta (TNF-β), induce IL-12 secretion from dendritic cells, and activate macrophages and CD8 T cells to eliminate intracellular pathogens. Upon activation, CD8 T cells, often with the help of Th1 cells supplying IFN-γ to upregulate B7 expression on APCs, become functional cytotoxic T killer cells. Conversely, Th2 cells elaborate IL-4, IL-5, IL-6, IL-10, and IL-13, which aids antibody and eosinophil production. There is also crosstalk between Th1 and Th2 cells; for example, IFN-γ from Th1 cells suppresses Th2 cells, and IL-10 from Th2 cells inhibits Th1 cells.
Tregs are a subset of T cells, which play a critical role in suppressing the activity of effector T cells that escape negative selection to self-antigens in the thymus. Functional Tregs are able to anergize previously self-reactive T-effector cells, resulting in improved tolerance to self. Tregs originating from the thymus are termed as central or natural Tregs and carry surface CD4 + CD25 + and intracellular forkhead transcription factor (FOXP3 + ) markers, which are specific to CD4 + CD25 + Treg cell population. FOXP3 is a transcription factor that is required for developing α/β TCR-positive T cells to differentiate into Tregs in the thymus. First identified in the Scurfy mouse, a mouse model of immune dysfunction and polyendocrinopathy, abnormal FOXP3 expression is now known to be responsible for failure in immune tolerance in humans affected with a similar polyendocrinopathy, as further discussed later. Abnormal FOXP3 expression in humans leads to an extremely rare, X-linked inherited, and typically fatal autoimmune lymphoproliferative disease known as IPEX (Immune dysregulation, Polyendocrinopathy, Enteropathy, and X-linked inheritance). Defects in FOXP3 responsible for IPEX map to Xp11.23-Xq13.3.
B-Cell Tolerance
Naïve B cells, during the early stage of development in the bone marrow, express surface immunoglobulin (Ig)M, which serves as B-cell receptors (BCRs). Upon interacting with self-antigens, naïve immature B cells undergo negative selection, either through clonal deletion or anergy, whereby B cells enter a state of unresponsiveness and have a reduced life span. Another process mediating B-cell central tolerance is receptor editing, whereby genetic rearrangement of the Ig chain leads to generation of BCRs with new antigen specificities. B cells with nonautoreactive BCRs are positively selected and continue to the periphery. If self-reactive B cells escape into the periphery, they undergo anergy. Anergized B cells do not die immediately but have a shorter half-life. Naïve mature B cells require T-cell help for realization of their full potential through affinity maturation and class switching. The absence of T-cell help also leads to B-cell tolerance.
Autoimmune Diseases
The organ-specific nature of many autoimmune diseases results from abnormal immune system recognition of tissue-specific self-antigens. In many autoimmune endocrinopathies, the target molecule is either a tissue-specific or tissue-limited (i.e., the protein is not unique to one tissue but is clearly restricted in its distribution) enzyme or cell-surface receptor ( Table 22.2 ).
| Disease | Autoantigens | Putative Autoantigens |
|---|---|---|
| Mucocutaneous Candidiasis | IL-17A IL-17F IL-22 | |
| Hypoparathyroidism | NALP5 CASR | |
| Addison disease | P450c21 | P450c17 P450scc |
| Hashimoto thyroiditis | Thyroid peroxidase Thyroglobulin | |
| Graves disease | Thyrotropin receptor | |
| Diabetes | Insulin Glutamic acid Decarboxylase 65 IA-2 (ICA 512) IA-2β ZnT8 | Proinsulin Carboxypeptidase H ICA69 Glima 38 |
| Premature gonadal failure | P450scc | P450c17 3 β hydroxysteroid dehydrogenase |
| Pernicious anemia | H + /K + ATPase pump Intrinsic factor | |
| Myasthenia gravis | Acetylcholine receptor α chain | |
| Vitiligo | Tyrosinase Tyrosinase-related protein 2 L-amino acid decarboxylase | |
| Celiac disease | Endomysium transglutaminase | Reticulin Deamidate gliadin |
| Autoimmune hepatitis | Liver kidney microsome 1 | L-amino acid decarboxylase Tryptophan hydroxylase |
The criteria for classification of a disease as autoimmune are not universally agreed upon. However, major criteria that are generally accepted as strong evidence of autoimmune disease include: (1) detection of autoantibodies or autoreactive T cells, including lymphocytic infiltration of the targeted tissue or organ; (2) disease transfer with antibodies or T cells; (3) disease recurrence in transplanted tissue; and (4) ability to abrogate the disease process with immunosuppression or immunomodulation. Few, if any, human autoimmune diseases meet all these criteria. Further information that is supportive of, but not diagnostic for an autoimmune disease, include: (1) increased disease frequency in women compared with men, (2) the presence of other organ-specific autoimmune diseases in affected individuals, and (3) increased frequencies of particular HLA alleles in affected individuals.
Defects in Tolerance That Cause Autoimmune Disease
Several different hypotheses explaining defects in tolerance have been proposed. Autoimmunity may develop because (1) tolerance never developed to specific self-antigens or (2) established tolerance was lost ( Fig. 22.3 ). If self-antigen is not efficiently presented in the thymus, tolerance may not be established during T-cell education within the thymic cortex. For example, AIRE mutations lead to lack of expression of ectopic self-antigens by mTECs and their presentation to developing T cells. This leads to escape of autoreactive T cells into the periphery and ultimately multiorgan autoimmunity. Another example is the insulin gene ( INS ) VNTR (variable number of tandem repeats), which lies around 500 basepairs upstream of the INS gene promoter. The INS VNTR influences thymic insulin T-cell expression and education based on its length. Specifically, longer VNTRs are associated with increased thymic expression of insulin, and thus a decreased risk of developing type 1 diabetes, whereas shorter VNTR are associated with decreased thymic expression of insulin, failure to delete specific autoreactive T-cell clones, and an increased risk of developing diabetes.
If tolerance has not developed because of intracellular sequestration of an antigen, and thus not expressed in the thymus during T-cell ontogeny, T-cell reactivity in the periphery will not be abolished. However, several antigens initially thought to be sequestered intracellularly have now been shown to circulate in low concentrations in normal individuals. Thyroglobulin, a self-antigen in autoimmune thyroid disease, is known to circulate in low but appreciable quantities in individuals with no serologic evidence of thyroid autoimmunity. Thyroid follicular cell destruction in Hashimoto thyroiditis is mostly cell mediated and not mediated by humoral factors.
If sequestered antigens do play a role in autoimmune disease, viral infections, trauma, ischemia, or irradiation are all possible mechanisms that could disturb cellular integrity and lead to release of intracellular antigens. Some sequestered self-antigens may never encounter the immune system, unless there is a breakdown of anatomic barriers within the body. An example is the occurrence of autoimmunity to intraocular proteins, following orbital trauma. Although a rare consequence of orbital damage, initiation of an autoimmune response to released sequestered intraorbital proteins in adjacent lymph nodes can generate autoreactive T cells that can invade and damage the contralateral eye (sympathetic ophthalmia). Removal of the inciting damaged tissues and immunosuppression may be required to sustain vision in the undamaged eye. Similarly, transient autoantibody reactivity to cardiac myosin following myocardial infarction has been described albeit with no pathological consequences.
Alteration of self-antigens because of infection or neoplasia is another possible theory explaining some types of autoimmunity. As environmental triggers, viral infections could lead to modification of self-proteins and neoantigen expression. Alternatively, a self-antigen may be partially degraded, leading to a “new” antigenic target for the adaptive immune system. This new antigen is recognized as foreign by the immune system, and the immune response to these new antigens results in autoimmunity. Some cells/tissues may suffer unintended autoimmune damage when substances bind to the cells and elicit an initial immune response. For example, certain drugs bind to red blood cells and result in an immune hemolytic anemia. If an antibody response to the red-cell–bound drug is elicited, the antigen-antibody complex present on the red blood cell can lead to red blood cell destruction. This can occur either through red blood cell phagocytosis by the monocyte-macrophage system or via complement-mediated lysis of the red blood cell. Thus the red blood cell becomes an innocent bystander to the antidrug humoral immune response. Theoretically, this could also occur with viruses that serendipitously attach to tissues.
Molecular mimicry is another mechanism to explain development of autoimmunity. Following exposure to a dietary, viral, or bacterial antigen (e.g., infection) and similarity (molecular mimicry) between the self-antigen and the foreign antigen, the immune response to the foreign antigen leads to cross-reactivity with self-antigen, autoimmunity, and disease. For this theory to work, tolerance must not previously exist to the self-antigen. This might be true if the self-antigen is truly sequestered and the immune system has never developed tolerance to the self-antigen. Alternatively, the self-antigen peptides may be present in very low concentration to elicit an immune response and initial tolerogenicity has not occurred. Only after infection or novel dietary exposure would there be a sufficient degree of immunization to the exogenous antigen (which is similar to a self-antigen) and subsequent immune autoreactivity. If the self-antigen is a cell-surface antigen, the “pathogen-induced” autoantibodies could fix to self and produce disease via complement fixation, or the antibodies could act as opsonins for fixed or circulating phagocytes (antibody-dependent cell cytotoxicity). In rheumatic fever, associated comorbidities such as carditis and Sydenham chorea are thought to be autoimmune manifestations secondary to similarities in the structural components of group A Streptococcus with collagen (I and IV) and fibronectin in human cardiac connective tissues and tubulin in human brain cells, respectively.
Some cases of autoimmunity may result from superantigens, which can be secreted by certain pathogenic bacteria and viruses. Superantigens are polyclonal T-cell stimulators that can cross-link TCR β chains and MHC molecules, and activate as many as a third of T cells in the body. This can initiate a nonspecific T-cell immune response, including against self-antigens. In such cases, systemic disease can develop from massive cytokine release (e.g., systemic inflammatory response syndrome [SIRS]). This is the case in toxic shock syndrome, wherein a staphylococcal exotoxin acts as a superantigen. Mycobacterial antigens have also been proposed as possible superantigens in Crohn disease. This theory requires that T cells bearing antiself TCRs have not been deleted or become permanently anergic. These T cells may be stimulated and proliferate to develop an autoimmune response if they encounter the specific self-antigen.
Similar to polyclonal T-cell activation, polyclonal B-cell stimulation has also been implicated in humoral autoimmunity. Autoreactive B cells arise routinely as part of the naïve B-cell repertoire, and can be found in healthy individuals. If an autoreactive clone of B cells encounters a self-antigen and a costimulator (which might be nonspecific, e.g., a virus, such as Epstein-Barr virus, or a bacterial product, such as lipopolysaccharide), autoantibodies could be produced, bypassing the need for T-cell help.
Autoimmune human disease likely results from an interaction of environmental and genetic factors. Environmental factors implicated include: wheat gliadin ingestion and celiac disease, penicillamine exposure and myasthenia gravis, methimazole and autoimmune hypoglycemia from insulin autoantibodies (reported primarily in Japanese patients), and amiodarone and thyroiditis. Cancer has also been associated with the development of autoimmunity: thymoma and myasthenia gravis, ovarian teratoma and N -methyl- D -aspartate receptor–meditated encephalitis, and breast cancer and stiff-person syndrome. Despite remarkable improvements in our understanding of immunology, the mechanisms whereby the complex interaction of genes, environment, and immune system lead to autoimmunity remain to be fully elucidated.
Checkpoint Inhibitors and Autoimmunity
Tumor cells can manipulate the inherent immune tolerance mechanism to disrupt antitumor immunity. An efficient way of escaping antitumor activity is by increasing checkpoint pathways, which suppress T-cell responses. An example is the interaction of CTLA-4 (present on activated T-cells) with B7.1/B7.2 of APCs that downregulates T cell response. CTLA-4 can also remove B7 molecules from APCs, through a process called transendocytosis and prevent binding of CD28 costimulatory molecules, and thus bring about T-cell anergy. Another example is the interaction of PD-1 on T cells with its ligands PD-L1 and PD-L2. PD-1/PD-L1/PD-L2 interactions inhibit T-cell proliferation and production of proinflammatory cytokines (TNF-α, IFN-γ, and IL-2), allowing immune checkpoint pathways to promote a tolerogenic environment.
Checkpoint inhibitors, now increasingly used in anticancer therapy, include monoclonal antibodies targeting the CTLA-4 and PD-1 pathway (both the PD-1 receptor and PD-L1 ligand) and thus removing the restraint on antitumor activity ( Fig. 22.4 ). Blockade of CTLA-4 enhances costimulatory signals and leads to naïve T cells having increased effector T-cell responses to tumor cells, whereas PD-1 pathway blockade leads to increased T-cell proliferation and a proinflammatory milieu that aids in antitumor activity. In addition, there are other immune pathways impacted by blockade of these pathways. The discovery of immune checkpoint inhibitors was a breakthrough in anticancer therapy, resulting in the 2018 Nobel Prize for Medicine awarded to Drs. James Allison and Tasuku Honzo, two pioneers in this field.
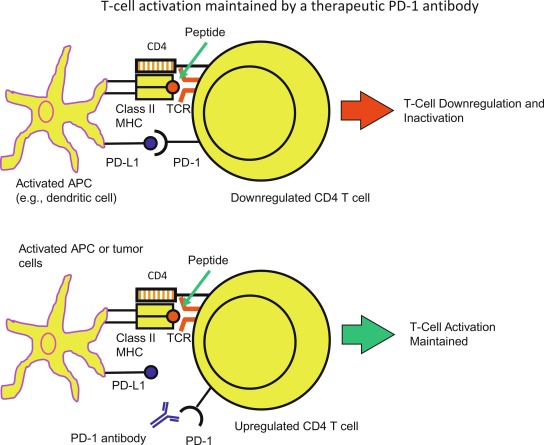
Given that checkpoint inhibitors have the ability to eliminate a tolerogenic environment, it is not surprising that these immunomodulatory agents can bring about immune-related adverse effects (irAEs), and such irAEs have even been referred to as the Achilles heel of cancer immunotherapy . Hypophysitis, hepatitis, dermatitis, colitis, type 1 diabetes, thyroiditis, adrenalitis, and myocarditis have been reported following treatment with ipilimumab (CTLA-4 antibody) and/or PD-1 antibody (nivolumab) therapy. Other potential mechanisms leading to irAES after use of checkpoint inhibitors include: (1) cross-presentation, where tumor antigens released after antitumor activity are picked up by APCs, initiating secondary immune responses, and (2) epitope spreading (after release of tumor antigens), whereby there is continuous acquisition of neo antigens and recruitment of untargeted T cells. Despite the success of checkpoint inhibitors as anticancer drugs, these irAEs remain a concern. There are few studies in cancer patients with and without preexisting autoimmune conditions, and the long-term effects of checkpoint inhibitor use remain unknown. Newer modes of anticancer immunotherapy that have been investigated to lessen irAEs include increasing the efficacy and the use of vaccines against tumor neoantigens.
Having provided a review of basic immunological concepts related to central and peripheral tolerance, we will shift our focus for the remainder of the chapter toward the clinical and pathological aspects of APS.
Classification of the autoimmune polyglandular syndromes
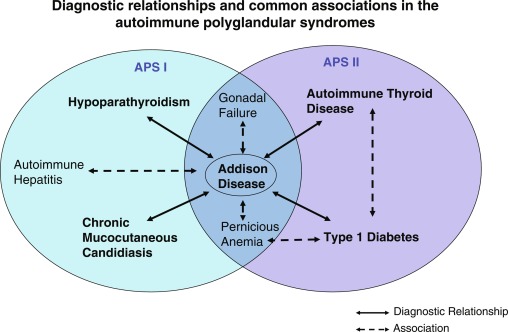
APS I, also known as a utoimmune p oly e ndocrinopathy c andidiasis- e ctodermal d ystrophy ( APECED ) is an autosomal recessive disorder characterized by several autoimmune disorders with significant heterogeneity in its presentation. The syndrome is caused by a mutation in the AIRE gene on chromosome 21q22.3. The presence of two of the following three conditions are prerequisites for the diagnosis of APS I: (1) adrenocortical failure (Addison disease) or serologic evidence of adrenalitis (adrenal autoantibodies), (2) hypoparathyroidism, and (3) chronic mucocutaneous candidiasis. APS II is defined by the coexistence of at least two of these three endocrinopathies: (1) autoimmune adrenocortical insufficiency or serologic evidence of adrenalitis, (2) autoimmune thyroiditis, and/or (3) type 1 diabetes mellitus or serological evidence of islet autoimmunity. Traditionally, the presentation of autoimmune thyroid and adrenal disease has been termed Schmidt syndrome , whereas a complete triad of adrenal, thyroid, and islet autoimmunity (or type 1 diabetes mellitus) has been termed Carpenter syndrome ( Fig. 22.5 ). The presence of thyroiditis, without adrenal disease, but associated with type 1 diabetes, pernicious anemia, vitiligo, or alopecia has been referred to by some authors as APS III , whereas additional combinations of autoimmune disease have been referred to as APS IV (i.e., vitiligo plus alopecia, type 1 diabetes plus celiac disease, or type 1 diabetes and vitiligo). Given that they share similar susceptibility genes and immunological features, these subtypes are often considered as extensions of APS II and not necessarily as separate entities.
Genetics of APS I and APS II
APS I is caused by mutations in the AIRE gene, and is inherited in an autosomal recessive fashion. The AIRE protein is expressed in the thymus, lymph nodes, and fetal liver, as well as in pancreas, adrenal cortex, and testes. The gene spans 11.9 kb, contains 14 exons, and encodes a 545 amino acid protein. The protein AIRE has four major domains—CARD (caspase recruitment domain), SAND ( S P100, A IRE1, N ucP41/ P75 and D EAF1), PHD1 (plant homeodomain), and PHD2. The CARD domain is involved in multimerization of the AIRE protein into an active state, and its attachment to a target chromatin. The SAND domain is required for AIRE to interact with a transcriptional repressive complex (ATF7ip), which in turn is critical for AIRE-dependent gene expression. The PHD domains function as sites for multiple binding events and aids AIRE transcriptional activity.
Over 98% of patients with APS I have mutations in the AIRE gene. So far, more than 100 mutations in the AIRE gene have been reported, and recessive mutations causing disease are distributed throughout the AIRE gene. Although the inheritance is traditionally accepted as autosomal recessive, there are some patients reported with heterozygous dominant mutations (in the gene encoding the SAND domain) and atypical or nonclassical manifestations of APS I. Because AIRE is active in multimeric forms, minor changes in amino acid sequences in critical domains of the AIRE secondary to heterozygous mutations can lead to defective multimers, and thus a negative dominant effect, whereby the altered gene product inhibits the functioning of the wild-type protein. Such an effect has also been reported for monoallelic mutations in the AIRE gene encoding the PHD1 domain.
Unlike APS I, APS II is not inherited as a single gene mutation but displays polygenic inheritance. APS II is much more typical of other autoimmune endocrinopathies, where cases can occur either sporadically, or within families with APS II. Often, different organ-specific manifestations share common genetic associations, and most of these genes code for key regulatory elements in immune system pathways. The MHC ( HLA in humans) class II complex involved in antigen presentation is the most important gene associated with APS II.
A study in 1986 reported an association of HLA-DR3 and/or DR4 haplotypes with Addison disease and type 1 diabetes. Subsequent studies have shown that variants in DR3-DQ2 and DR4-DQ8 haplotypes confer a risk for type 1 diabetes and Addison disease, as well as autoimmune thyroid disease and celiac disease. This may indicate a common immunogenetic etiopathogenesis and highlights why multiple autoimmune disorders can develop in the same person. Other components of APS II, such as Graves disease, have shown association with HLA-DR3 as opposed to Hashimoto thyroiditis, which is associated with HLA-DR4 or DR5 . In addition to class II molecules, class I molecules ( HLA-A and B ) have also been implicated in the risk for type 1 diabetes.
Because the association of APS II with specific HLA alleles is only modest, the role of other susceptibility genes has been studied. Other non- HLA genes conferring APS II risk include genes that encode CTLA-4, the transcriptional regulator protein BACH2, protein tyrosine phosphatase nonreceptor type 22 (PTPN22), and CD25 (the high-affinity IL-2 receptor).
Clinical aspects
The major disease components, frequencies, and differences between APS I and II are shown in Table 22.1 . The overlap in some disease components between APS I and II are also highlighted in Fig. 22.5 .
APS I
Although the disease is not common, with an overall prevalence of less than 1:100,000, it is more prevalent in Finland (1:25,000), Sardinia (1:14,000), and among Iranian Jews in Israel (1:9000). The Finnish cohort of 91 subjects is the largest and most well-characterized APS I group of patients worldwide.
Persistent mucocutaneous candidiasis is usually the first sign (60% of all APS I patients) and appears during the first year or two of life. In the Finnish cohort, 50% developed candidiasis by age 5 years, 94% by age 20 years, and 100% by 40 years of age. Candidal diaper rash is observed early in life, with vulvovaginal candidiasis often developing at puberty in females. Colonization of the gut by candida can lead to intermittent abdominal pain and diarrhea. Retrosternal pain in patients with oral candidiasis may suggest esophageal candidiasis and should be confirmed by esophagoscopy. Infection of the nails with chronic candidiasis may lead to a darkened discoloration, thickening, or erosion. Studies have shown that T cells from AIRE-deficient individuals still have a competent proliferative response against C. albicans . However, there is defective receptor-mediated internalization of yeast cell wall derivatives by monocytes in AIRE-deficient antigen presentation, and this leads to ineffective clearing of mucocutaneous candidiasis, and thus the chronicity. Thus, all patients with refractory mucocutaneous candidiasis should be thoroughly investigated not only for a T-lymphocyte abnormality (absolute lymphocyte count, enumeration of T-cell subpopulations, assessment of T-cell function where possible) but also for the presence of polyendocrinopathies. Chronic oral mucosal candidiasis must be treated aggressively, as there is increased risk of candidiasis associated squamous cell carcinoma (SCC) of the oral mucosa or esophagus; SCC was reported in seven of the 55 Finnish APS I patients over 25 years of age, albeit five of the seven were also smokers.
Oral mucous membranes must be protected from exposures that can increase susceptibility to candida infections. Specifically, patients should be advised to avoid hard, sharp, or spicy foods, as well as whitening toothpastes or abrasives. Dentures or orthodontics can provide additional surfaces for candida growth. Fluconazole has good activity against candida and is the preferred treatment. Other azoles that have been used include ketoconazole, and miconazole. Data suggest that prolonged use can lead to resistance. With drug resistance, newer azoles, such as itraconazole, voriconazole, or posaconazole can be used. Patients should follow up closely with an infectious disease specialist. Physicians must be cognizant of the possibility of precipitating adrenal insufficiency or worsening already present adrenal deficiency when using ketoconazole; ketoconazole can inhibit cortisol synthesis. In addition, there have been reports of adrenal insufficiency following use of other azoles (fluconazole, posaconazole) in sick patients.
Hypoparathyroidism is typically the first endocrinopathy to develop in APS I and eventually occurs in more than 85% of patients. Hypoparathyroidism usually presents after the onset of mucocutaneous candidiasis but before puberty, with 33% of APS I patients diagnosed by age 5 years, 66% by age 10 years, and nearly 85% by age 30 years. Severe hypocalcemia, as evidenced by seizures, carpopedal spasms, muscle twitching, and laryngospasm may be presenting features of APS I, although these symptoms may be masked by the relative hypercalcemia associated with the cooccurrence of adrenal insufficiency. Some of the theories to explain the development of hypercalcemia in adrenal insufficiency include: (1) hypovolemia-induced reduction in glomerular filtration, and hence the amount of calcium filtered through the glomerulus, and (2) increased activity of 1α hydroxylase, which is normally inhibited by glucocorticoids. Low or inappropriately normal intact parathyroid hormone (PTH) with concurrent hypocalcemia and hyperphosphatemia are diagnostic of hypoparathyroidism. Standard therapy consists of oral calcium salts and calcitriol administration (see Chapter 20 for management). Whereas recent data suggest that once or twice daily administration of subcutaneous recombinant parathyroid hormone (rPTH 1-34 and rPTH 1-84) may provide optimal therapy, especially in those who are poorly controlled on standard therapy, this approach is not yet approved by the US Food and Drug Administration for children in the United States.
Autoimmune adrenocortical insufficiency (Addison disease) is the third major component of APS I and typically occurs after mucocutaneous candidiasis and hypoparathyroidism have been diagnosed. Over 85% of APS I patients eventually develop adrenal insufficiency. Unfortunately, the clinical diagnosis of adrenal insufficiency is often missed initially with the diagnosis commonly made late, or at the time of a life-threatening adrenal crisis. In the Finnish cohort of APS I patients, 40% had Addison disease by 10 years of age and nearly 80% by 30 years of age. Deficiencies of cortisol, aldosterone, and adrenal androgens may present simultaneously or may evolve over months to years. In addition, the initial symptoms of adrenal insufficiency are often nonspecific, mimicking psychiatric or gastrointestinal disease. These include fatigue, weight loss, myalgias, arthralgias, behavioral changes, nausea and vomiting, abdominal pain, and diarrhea. Cortisol deficiency leads to increased pituitary production of proopiomelanocortin or POMC, a precursor molecule that is cleaved into products including adrenocorticotrophic hormone (ACTH) and melanocyte-stimulating hormone (MSH). Over time, hyperpigmentation (because of elevated MSH) in nonsun-exposed areas, along with postural hypotension, can usually be found on careful examination. Unexplained hypotonic dehydration, with concomitant presence of the mentioned features, should raise the suspicion of Addison disease. Adrenal crisis with hyponatremia, hyperkalemia, acidosis, hypotension, and hypoglycemia may be fatal, unless recognized and treated appropriately and promptly with intravenous glucocorticoids and isotonic fluids (please see specific chapters for more details). As discussed in detail further in the chapter, adrenal autoantibodies are used to predict adrenal cortical failure. If antibody testing is positive, morning cortisol and renin measurements and/or an ACTH stimulation testing may be used diagnostically or monitoring asymptomatic patients.
Autoimmune gonadal failure occurs in over 50% to 60% of women with APS I by age 20 years, whereas less than 25% of males develop testicular insufficiency. Gonadal failure often presents with primary amenorrhea in young women though menstrual irregularities, polycystic ovaries, or infertility may be presenting features. As with autoimmune adrenalitis, gonadal failure can be predicted by the presence of steroidal cell autoantibodies.
Ectodermal dystrophy, unrelated to hypoparathyroidism or mucocutaneous candidiasis, has been extensively documented in the Finnish cohort. Dental enamel hypoplasia of permanent (but not deciduous) teeth, as well as nail dystrophy (nail pits of 0.5–1 mm), are commonly found. There may be complete absence of the enamel or transverse hypoplastic bands alternating with zones of well-formed enamel. Nearly one-third of the Finnish patients also had calcification of the tympanic membranes and 20% to 25% developed keratitis.
As shown in Table 22.1 , type 1 diabetes and thyrogastric autoimmunity (a descriptive term for the combination of autoimmune thyroid disease and atrophic gastritis) are associated with APS I, but occur far less frequently than in APS II. When present, thyroiditis is typically atrophic rather than goitrous. Gastric-parietal cell autoimmunity, which leads to atrophic gastritis, with resultant achlorhydria and intrinsic factor deficiency, typically presents as iron-deficiency or vitamin B 12 -deficient pernicious anemia. Atrophic gastritis occurs in 15% to 30% of APS I cases with a mean age of onset of 16 years. Whereas iron-deficiency anemia is microcytic and vitamin B 12 -deficient anemia is macrocytic, combined iron and vitamin B 12 deficiency can be normocytic. It is also important to recognize that the spinal cord comorbidities (subacute combined degeneration of the spinal cord) of vitamin B 12 deficiency can occur in the absence of anemia.
Nonendocrine organ-specific manifestations are less common (~ 5%–20%) and include alopecia, vitiligo, autoimmune hepatitis, and malabsorption among others. Progression to alopecia totalis (total loss of scalp hair) or universalis (total loss of all body hair, including eyelashes, eyebrows, and scalp hair) usually occurs before puberty. Vitiligo presents initially as small, pale pigment-lacking skin patches. These may be missed, unless specifically sought and ultraviolet light examination of the skin may be necessary. The appearance of clay-colored stools, dark urine, and jaundice suggests chronic active autoimmune hepatitis. Hepatitis occurs in 10% to 15% of APS I patients and is the leading cause of death. Consequently, all patients suspected of having APS I should have their liver function regularly monitored. Autoimmune hepatitis is typically treated with glucocorticoids initially, and later with azathioprine, once there is partial disease control. Malabsorption, which may occur intermittently (and typically of fat), has been linked to hypoparathyroidism (insufficient duodenal mucosal hormone and pancreatic enzymes because of hypocalcemia), bacterial and fungal overgrowth, gluten sensitivity (celiac disease), and IgA deficiency. There have also been rare reports of APS I with hypophysitis (diabetes insipidus, growth hormone deficiency, ACTH deficiency), and nonendocrine manifestations, such as rheumatoid arthritis, Sjögren disease, tubulointerstitial nephritis, autoimmune bronchiolitis, myopathy, and asplenia. Recent studies in a European cohort of 112 APS I patients have also uncovered a wide spectrum of phenotypic manifestations and genotypic changes in AIRE.
A study in a cohort of 35 North and South American subjects revealed that nonendocrine manifestations occur much earlier and more frequently when compared with European cohorts. These nonendocrine manifestations included urticarial eruptions (66%), hepatitis (43%), gastritis (48%), intestinal dysfunction (80%), Sjögren-like syndrome (43%), and pneumonitis (40%), and were present in 80% before the diagnostic dyad criteria of APS were met. It is unclear if there are genotype-phenotype correlations that can explain the higher frequency of nonendocrine manifestations in the American cohort.
APS II
APS II is the most common of the autoimmune polyendocrinopathies (excluding the coincidences of type 1 diabetes and autoimmune thyroid disease), with a prevalence of 1 to 4:100,000 and with a polygenic inheritance. Unlike APS I, APS II usually has its onset in adulthood, particularly during the third or fourth decade and is at least 3 times more common in females than males, whereas APS I is equally common in males and females. This female bias in APS II is primarily explained by the coexistence of autoimmune thyroid disease (AITD), which is more common in women. In 1926 Schmidt first described the association of adrenocortical and thyroid gland failure and Carpenter extended this in 1964 to include type 1 diabetes mellitus. In 1957 the autoimmune nature of these diseases was suggested by Doniach and Roitt’s discovery of thyroglobulin autoantibodies in patients with Hashimoto thyroiditis.
APS II presents with adrenocortical failure in approximately 50% of cases, though multiple disease components may be present at diagnosis. Although the disease usually has its onset between ages 20 and 50 years, it is not unusual to find cases before or after these ages. Type 1 diabetes coexists in nearly 50% of patients with Addison disease, whereas AITD coexists in about two-thirds of patients with Addison disease. Therefore type 1 diabetes and AITD must be pursued vigorously in any patient presenting with Addison disease.
The most common component of APS II is AITD and is seen in almost 70% to 75% of patients. AITD affects nearly 4.5% of the US population, with 80% to 90% of all cases occurring in females. AITD increases in incidence during the teen years, peaking in the fifth and sixth decades. Chronic lymphocytic thyroiditis (Hashimoto disease) is by far the most common form of AITD, although Graves disease may also occur. Postpartum thyroiditis has also been considered a transient manifestation of autoimmune thyroiditis following childbirth and can manifest as hypo- or hyperthyroidism. Several studies have reported on the coexistence of islet autoimmunity (3%–8%) or even overt type 1 diabetes and AITD, whereas less than 1% of patients, with otherwise isolated thyroiditis, have serological evidence of adrenal autoimmunity.
Although “polyglandular syndrome” involvement in patients with autoimmune thyroid disease is infrequent, thyroid autoimmunity or a family history of thyroiditis is common in patients with pernicious anemia, vitiligo, alopecia, myasthenia gravis, and Sjögren syndrome. Almost 20% to 40% of vitiligo patients have another component of APS II, with thyrogastric autoimmunity being the most common. Most patients with vitiligo are asymptomatic and evidence of concurrent autoimmunity can be ascertained only by autoantibody screening. Segmental vitiligo with involvement of dermatomal regions is not associated with autoimmunity. Up to 15% of patients with alopecia (areata, totalis, universalis) and 5% of their first-degree relatives have thyroid disease. More patients with APS I than APS II have cutaneous manifestations, such as vitiligo or alopecia, but because APS II is far more common, most patients with either of the manifestations and another autoimmune disease are categorized as incomplete APS II.
Nearly 30% of patients with myasthenia gravis, an autoimmune disease characterized by presence of anti-acetylcholine receptor autoantibodies and muscle weakness worsening during muscular contraction, have AITD. Both Hashimoto thyroiditis and Graves disease may occur in patients with myasthenia gravis. Interestingly, patients with myasthenia gravis and concomitant AITD tend to have milder expression of their myasthenia, and a lower incidence of thymic disease and acetylcholine receptor α-chain autoantibodies. The incidence of ocular myasthenia is higher in patients with Graves disease.
Type 1 diabetes mellitus, a diagnostic component of APS II, has a peak incidence during the teen years with a smaller but increasing incidence occurring in the preschool years. Nevertheless, the disease may have its onset at any age. Approximately 10% to 15% of APS patients, incorrectly labeled as type 2 diabetes because of diabetes onset after 40 years of age, actually have slowly progressive autoimmune diabetes (also referred to as latent autoimmune diabetes of adults or LADA). Unlike APS II, in which there is a female gender bias despite the occurrence of type 1 diabetes, no gender bias is present in patients with isolated type 1 diabetes. AITD (denoted by the presence of thyroperoxidase and/or thyroglobulin autoantibodies) occurs in 20% to 25% of patients with type 1 diabetes with women representing nearly two-thirds of the autoantibody-positive patients. Despite the high prevalence of thyroid autoantibodies, less than 20% of patients with thyroid autoantibodies have evidence of thyroid dysfunction defined as an elevated thyroid-stimulating hormone (TSH) concentration. Adrenocortical autoimmunity is much less frequent among patients with type 1 diabetes, with serological evidence reported in 1.5% of cases. Tissue transglutaminase autoantibodies suggestive of celiac disease are present in 3% to 7% patients with type 1 diabetes. Celiac disease should be suspected in type 1 diabetes patients with unexplained diarrhea, weight loss, failure to gain weight, or failure to thrive, unexplained hypoglycemia should be confirmed by intestinal biopsy.
Gastric parietal cell autoantibodies (PCAs) are present in approximately 10% of females and 5% of males with type 1 diabetes. Although pernicious anemia typically affects women after the fifth decade, children with PCA should be monitored closely for the development of pernicious anemia. Atrophic gastritis may lead to the development of megaloblastic anemia because of lack of intrinsic factor required for absorption of dietary vitamin B 12 from the gut. Iron-deficiency anemia may also occur in both adolescents and adults because of impaired ability to absorb iron consequent to decreased acid production (achlorhydria).
Approximately 10% of women younger than 40 years of age with APS II develop ovarian failure. Ovarian failure may present as either primary or secondary amenorrhea. In females with biopsy-proven lymphocytic oophoritis, adrenocortical failure or subclinical adrenal autoimmunity is often present. In contrast, progression to gonadal failure is very rare among males with Addison disease.
Pituitary involvement is occasionally seen in APS II. Hypophysitis and empty sella syndrome have been described, leading to isolated failure of secretion of growth hormone, ACTH, TSH, follicle-stimulating hormone (FSH), or luteinizing hormone (LH).
Several nonendocrinological conditions have also been reported in association with APS II. These include ulcerative colitis, primary biliary cirrhosis, sarcoidosis, achalasia, myositis, and neuropathy.
IPEX Syndrome
IPEX is a syndrome caused by defective Tregs secondary to mutations in the FOXP3 gene. The FOXP3 gene encodes a transcriptional factor of the same name. More than 70 FOXP3 mutations associated with IPEX have been described to date. Because FOXP3 is located in the X chromosome, the inheritance is X- linked recessive with males being affected, whereas females are carriers.
Patients who have FOXP3 inactivating mutations resulting in FOXP3 deficiency develop IPEX, which is characterized by multiorgan autoimmunity that begins very early in life and typically includes the triad of neonatal-onset type 1 diabetes, eczematous dermatitis, and enteropathy (watery diarrhea). Other autoimmune disorders include thyroiditis, hemolytic anemia, thrombocytopenia, hepatitis, nephropathy, arthritis, and lung disease. Patients with IPEX syndrome often manifest autoantibodies early in life; those with type 1 diabetes often have glutamic acid decarboxylase autoantibodies (GADA), and other islet cell autoantibodies (ICAs) in the neonatal period. Other autoantibodies detected early in life include those against harmonin and villin, proteins found in the microvilli of the intestinal brush border and the renal proximal tubule, and may explain the enteropathy and nephritis found in these patients.
IPEX is often fatal in the first few years of life. To date, only long-term immunosuppression or bone marrow transplantation, with the goal of enhancing regulatory T-cell function, have been effective therapies for IPEX. Sustained expression of FOXP3 may reprogram effector T cells to act as regulatory T cells. Novel approaches, including treatment with engineered Tregs or targeted gene editing of FOXP3 , are being investigated as treatment options for IPEX syndrome.
Diagnostic approach and follow-up
The approach to diagnosing polyglandular syndromes is threefold: (1) autoantibody screening to (i) verify the autoimmune nature of the suspected endocrinopathy, and (ii) test for the involvement of other organs and tissues; (2) full assessment of endocrine function in patients with confirmed autoantibodies and autoantibody-negative subjects in whom disease is suspected clinically; and (3) mutation analysis to confirm the diagnosis, and screen siblings and other relatives for their potential carrier status.
Recognition of multiorgan autoimmune diseases before their symptomatic phases is vital to minimize associated morbidity and mortality. A thorough history and physical examination should always be performed, and a high index of suspicion should be maintained. In addition, family history of multiorgan autoimmune disease should increase the suspicion for potential APS.
As discussed later, perhaps the greatest single achievement in the last decade was the discovery of the association of autoantibodies to type 1 interferons and APS I, enabling early diagnosis of APS I patients. Although not indicative of tissue-specific immune attack, autoantibodies to interferon (α and ω) provide a nearly 95% to 100% sensitive and specific screening test for APS I. The presence of autoantibodies to interferons should be followed by confirmatory testing for AIRE mutations, as well as testing for other tissue specific autoantibodies ( Fig. 22.6 ). These include 21-hydroxylase or adrenal cortex cytoplasmic autoantibodies (for autoimmune Addison disease), GADA, insulinoma associated autoantibodies (IA-2A), insulin autoantibodies (IAA), zinc transporter 8 (ZnT8) autoantibodies (for type 1 diabetes), thyroperoxidase and thyroglobulin autoantibodies (for AITD), steroidal cell autoantibodies (for ovarian failure), and transglutaminase autoantibodies (for celiac disease). In addition, although not readily available, autoantibodies to 17-hydroxylase, side-chain cleavage enzyme, and 3-hydroxysteroid dehydrogenase can be used to detect gonadal and adrenal autoimmunity. Autoantibodies to NACHT leucine-rich repeat protein 5 (NALP5) and calcium sensing receptor indicate underlying autoimmune hypoparathyroidism (CaSR). However, the specificity of CaSR and NALP5 for diagnosing autoimmune hypoparathyroidism is around 83% and 50%, respectively, whereas sensitivity has been reported to be 39% and 26%, respectively.