26 Aplastic anaemia (AA) may be part of a congenital syndrome, be secondary to well-defined insults to the bone marrow, or arise apparently spontaneously with no identifiable cause. A simple classification is shown in Table 26.1. The most common congenital disorder is Fanconi’s anaemia. Affected children suffer from defective DNA repair and the aplasia often coexists with skeletal deformities, skin pigmentation (Fig 26.1) and renal abnormalities. To date, fifteen genes (termed FANC) have been identified. Dyskeratosis congenita, another form of constitutional aplasia, is distinguished by a later onset, nail dystrophy, leukoplakia of mucosal surfaces and a high incidence of epithelial tumours. There is defective telomere maintenance and patients usually have very short telomeres. This is also observed in 10–15% of patients with acquired AA. Table 26.1 Classification of aplastic anaemia Fig 26.1 Fanconi’s anaemia. Infections known to predispose to AA include viral hepatitis and parvovirus infection. Exposure to chemicals, drugs and radiation can damage stem cells. Drugs may depress haematopoiesis idiosyncratically or predictably (Table 26.2). In roughly two-thirds of patients, no cause is apparent and AA is termed ‘idiopathic’. Improved haematopoiesis following immunosuppression (see below) suggests that in at least some cases the abnormal stem cell compartment is further compromised by poorly defined immune phenomena. Table 26.2 Drugs associated with aplastic anaemia1 1This is a selective list of more commonly implicated agents.
Aplastic anaemia
Classification and aetiology
1. Idiopathic AA
2. Congenital AA
Fanconi’s anaemia
Dyskeratosis congenita
3. Secondary AA
Drugs – idiosyncratic or dose-related
Chemicals
Ionising radiation
Infection
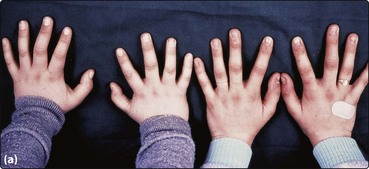
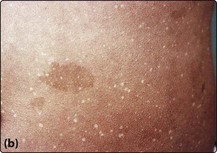
(a) Digital abnormalities in brothers with the syndrome. (b) Skin pigmentation.
Predictable
Cytotoxic agents
Idiosyncratic
Chloramphenicol
Sulfonamides
Phenylbutazone
Indometacin
Gold salts
Penicillamine
Carbamazepine
Phenytoin
Carbimazole
Oncohema Key
Fastest Oncology & Hematology Insight Engine



