As reviewed in other chapters, platelet activation and aggregation play a key role in atherothrombosis.1 Antiplatelet drugs target enzymes or receptors involved in the platelet synthesis of important agonists (e.g., thromboxane [TX] A2) or in mediating their activating/amplifying effects, respectively (FIGURE 111.1).2, 3 Because of the multifactorial nature of atherothrombosis, antiplatelet drugs can only avoid a fraction (typically, one-quarter to one-third) of major vascular complications. This protective effect against the occurrence of a first (primary prevention) or recurrent (secondary prevention) myocardial infarction or ischemic stroke cannot be dissociated from enhanced risk of bleeding complications, because of the important role played by the same drug targets in primary hemostasis. Moreover, antiplatelet therapy should be viewed within the context of a multifactorial approach to cardiovascular prevention that includes lifestyle modifications as well as other pharmacologic interventions.
This chapter reviews the pharmacologic basis and clinical evidence supporting the efficacy and safety of antiplatelet drugs for the treatment and prevention of atherothrombosis.
ASPIRIN
Aspirin (acetylsalicylic acid) was originally developed by the Bayer Company as an analgesic, antipyretic, and antiinflammatory agent at the end of the 19th century, and represented the prototypic nonsteroidal anti-inflammatory drug (NSAID). Following the discovery of its mechanism of action and characterization of its unique pharmacodynamic properties in inhibiting platelet function (reviewed in Ref.4), aspirin was developed as an antiplatelet agent during the past 30 years, a process largely driven by the medical-scientific community (reviewed in Ref.5).
Pharmacokinetics and Pharmacodynamics
Aspirin is rapidly absorbed in the stomach and upper intestine. Peak plasma levels occur 30 to 40 minutes after oral intake, and maximal inhibition of platelet function is achieved by 1 hour.2 In contrast, it can take up to 3 to 4 hours to reach peak plasma levels after the oral administration of enteric-coated aspirin. The oral bioavailability of regular aspirin tablets is approximately 50% over a wide range of doses,6 because of deacetylation of the drug during passage through the liver (first-pass effect). However, substantial variability has been reported in the oral bioavailability of enteric-coated aspirin formulations.7 Poor absorption from the higher pH environment of the small intestine and lower bioavailability of some enteric-coated aspirin preparations may result in inadequate platelet inhibition, particularly in heavier subjects.7
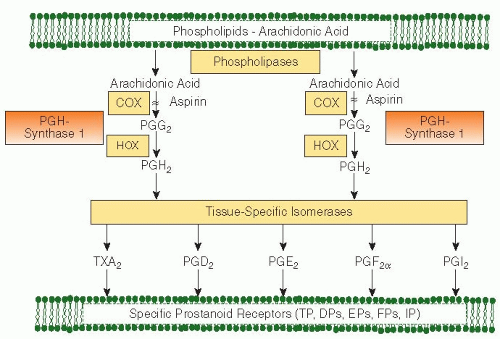
The plasma concentration of aspirin decays with a half-life of 15 to 20 minutes.2 Despite the rapid clearance of the intact drug moiety from the circulation, aspirin induces a long-lasting functional defect in human platelets. This appears to be primarily, if not exclusively, due to permanent inactivation by aspirin of a key enzyme in platelet arachidonate metabolism (FIGURE 111.2). This enzyme, prostaglandin (PG) H-synthase 1, is responsible for the formation of PGH2, the precursor of TXA2. In human platelets, TXA2 provides a mechanism for amplifying the activation signal by being synthesized and released in response to various platelet agonists (e.g., collagen, adenosine diphosphate [ADP], platelet-activating factor, thrombin) and, in turn, inducing irreversible aggregation.1
Aspirin selectively acetylates the hydroxyl group of a single serine residue at position 529 (Ser529) within the polypeptide chain of platelet PGH-synthase 1. This enzyme exhibits two distinct catalytic activities: a bis-oxygenase (cyclooxygenase [COX]) involved in formation of PGG2 and a hydroperoxidase (HOX) allowing a net two-electron reduction in the 15-hydroperoxyl group of PGG2, thus yielding PGH2(FIGURE 111.2). An inducible form of PGH-synthase has been identified and termed PGH-synthase 2 or COX-2.8 While newly formed platelets can express both COX-1 and COX-2,9 mature platelets primarily display COX-1 activity. As a consequence of O-acetylation of Ser529 by aspirin, the COX activity of the enzyme is lost permanently, whereas the HOX activity is not affected. The highly hydrophobic environment of the COX-1 channel stabilizes the aspirinmodified serine side chain against hydrolysis. As a result of this permanent modification of the enzyme, COX activity can only resume through new protein synthesis (occurring within a few hours in nucleated cells) or cell turnover (occurring at a daily rate of 10% to 12% in the case of human platelets).2 Aspirin is equally potent in acetylating COX-1 and COX-2, when tested in vitro.10 However, its unique mechanism of action and unusual pharmacokinetic features6 (short half-life and presystemic encounter with the platelet target in the portal blood prior to first-pass liver metabolism) allow selective, cumulative inhibition of platelet COX-1 at low doses while substantially sparing endothelial COX-2.2 Whereas the effect of aspirin on COX-1-dependent TXA2 production is saturable at daily doses as low as 30 mg,11 its inhibitory effect on COX-2-derived PGI2 production is dose dependent up to daily doses of 650 to 1,300 mg.12
The strikingly nonlinear relationship between inactivation of platelet COX-1 and inhibition of TXA2-dependent platelet function by low-dose aspirin (i.e., relatively limited inhibition of platelet function between 0% and 95% inhibition of COX-1; exponential increase in platelet inhibition between 97% and 100% COX-1 inactivation)13, 14 has important clinical implications: (a) a substantial loss of platelet inhibition is associated with less than maximal inactivation of COX-1; (b) recovery of platelet function is disproportionately rapid upon drug withdrawal; and (c) the requirement for virtually complete and persistent inhibition of platelet COX-1 cannot be met by most traditional NSAIDs, allowing their COX-2-dependent cardiotoxicity to be unmasked.15
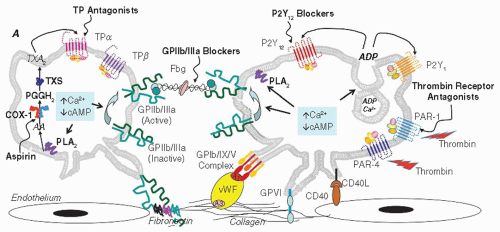
FIGURE 111.1 Platelet activation and aggregation: agonists, receptors, and site of action of antiplatelet drugs. The activation, adhesion, and aggregation of platelets are triggered by several agonists interacting with specific receptors on the platelet membrane. The GPIb/IX/V complex plays an essential role in the initial phase of platelet-vessel wall interaction that finally converges to GPIIb/IIIa activation. vWF binds to GPIb and collagen, while the subendothelial collagen binds to GPIa/IIa and GPVI as well. Platelet activation by agonists results in the decrease in cAMP and/or increase in intracellular Ca2+, leading to the inside-out change in the ligand-binding properties of GPIIb/IIIa, which conformationally switches to a high-affinity state for adhesive proteins such as fibrinogen, vWF, fibronectin, and vitronectin. Fibrinogen-mediated bridging of adjacent platelets via activated GPIIb/IIIa is the final mediator of aggregation. This response is amplified by further release of ADP and TXA2 and degranulation, which induce additional platelet activation/recruitment, changes in morphology and aggregation stability via outside-in signaling events mediated by activated GPIIb/IIIa. Antiplatelet drugs target enzymes (e.g., COX-1) or receptors (e.g., P2Y12) involved in the platelet synthesis of endogenous agonists (e.g., TXA2) or in mediating their activating/amplifying effects, respectively. ADP, adenosine diphosphate; cAMP, cyclic adenosine monophosphate; TX, thromboxane; TP, thromboxane receptor; PKC, protein kinase C; PAR, protease-activated receptor; TXS, thromboxane synthase; PGH2, prostaglandin H2; COX, cyclooxygenase; AA, arachidonic acid; PLA2, phospholipase A2; GP, glycoprotein; vWF, von Willebrand factor; CD40L, CD40 ligand; Fbg, fibrinogen.
FIGURE 111.2 Arachidonic acid metabolism through the PG H-synthase pathways. Arachidonic acid, a 20-carbon fatty acid containing four double bonds, is liberated from the sn2 position in membrane phospholipids by several forms of phospholipase A2, which are activated by diverse stimuli. Arachidonic acid is converted by cytosolic PGH-synthases, which have both COX and HOX activity, to the unstable intermediate PGH2. The synthases are colloquially termed COXs and exist in two forms, COX-1 and COX-2. Aspirin inhibits both COX-1 and COX-2. PGH2 is converted by tissue-specific isomerases to multiple prostanoids. These bioactive lipids activate specific cell-membrane receptors of the superfamily of G-protein-coupled receptors. TP denotes the TXA2 receptor, DP the PGD2 receptor, EP the PGE2 receptor, FP the PGF2α receptor, and IP the PGI2 receptor.
Efficacy and Safety
A very large database of randomized clinical trials (RCTs) now offers the most compelling evidence that prevention of myocardial infarction and ischemic stroke by aspirin is largely due to permanent inactivation of platelet COX-1.2, 5 These studies, which tested the efficacy and safety of the drug when given at daily doses ranging from as low as 30 mg to as high as 1,500 mg, have established two important facts. First, the antithrombotic effect of aspirin is saturable at doses in the range of 50 to 100 mg, as would be expected from human studies of platelet COX-1 inactivation.11 Second, despite a half-life of approximately 20 minutes in the human circulation, the antithrombotic effect of aspirin is observed with a dosing interval of 24 to 48 hours, reflecting the permanent nature of platelet COX-1 inactivation and the duration of TXA2 suppression following oral dosing in human.11 Other mechanisms of action that have been suggested to contribute to the antithrombotic effect of aspirin, such as a direct anti-inflammatory effect of the drug, are incompatible with these unique properties, because they require higher doses and shorter dosing interval.2
Aspirin’s unique feature in inhibiting platelet COX-1—its ability to inactivate the enzyme permanently through a short-lived active moiety—is ideally suited to its role as an antiplatelet drug, because it limits the extent and duration of extraplatelet effects of the drug, including the inhibition of PGI2.12 Moreover, the cumulative nature of platelet COX-1 acetylation by repeated low doses of aspirin11 explains the clinical efficacy of doses as low as 30 to 50 mg daily,2 the predictable high-grade inhibition of platelet TXA2 biosynthesis,14 and the persistence of the drug’s effect.2 These features, in turn, may limit the consequences of less-than-ideal compliance in a real world setting.
Permanent inactivation of platelet COX-1 by aspirin may lead to the prevention of atherothrombosis as well as to excess bleeding. At least two distinct COX-1-dependent mechanisms contribute to the increased risk of upper gastrointestinal (GI) bleeding associated with aspirin exposure: inhibition of TXA2-mediated platelet function and impairment of PGE2-mediated cytoprotection in the GI mucosa.2 Whereas the former effect is dose independent for daily doses >30 mg, the latter effect is clearly dose dependent.2 Inhibition of platelet function is largely responsible for the twofold increase in the risk of upper GI bleeding associated with daily doses of aspirin in the range of 75 to 100 mg, inasmuch as a similar relative risk (RR) is associated with other antiplatelet agents (e.g., clopidogrel) that do not act on COX and therefore do not affect PGE2-mediated cytoprotection.16 Inhibition of COX-1-dependent cytoprotection of the GI mucosa amplifies the risk of bleeding/perforation by causing new mucosal lesions or aggravating existing ones and is associated with a RR of four to six at the higher, analgesic, or anti-inflammatory doses of aspirin.2
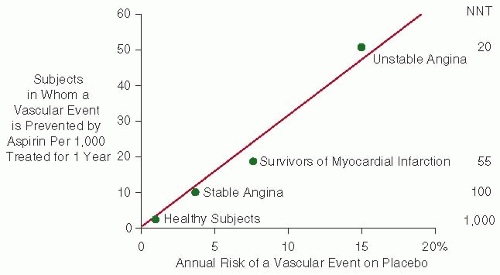
FIGURE 111.3 The risk of vascular complications is the major determinant of the absolute benefit of antiplatelet therapy. Data are plotted from placebo-controlled aspirin trials in different clinical settings. For each category of patients, the abscissa denotes the absolute risk of experiencing a major vascular event as recorded in the placebo arm of the trial(s). The absolute benefit of antiplatelet treatment is reported on the ordinate as the number of subjects in whom an important vascular event (nonfatal MI, nonfatal stroke, or vascular death) is actually prevented by treating 1,000 subjects with aspirin for 1 year. Numbers needed to treat (NNT) are also reported for each clinical setting.
The hypothesis that both the reduction in atherothrombotic complications and the increase in bleeding events are primarily due to inhibition of platelet TXA2 biosynthesis is supported by the recent finding that a highly selective TXA2 receptor (TP) antagonist, terutroban, and low-dose (100 mg daily) aspirin were associated with virtually identical rates of recurrent vascular events and major bleeds in an RCT of over 19,000 patients with a recent stroke.17 The possibility that aspirin may exert “pleiotropic” effects at low doses is not incompatible with a single mechanism of action, that is, inactivation of platelet COX-1, explaining antithrombotic, anti-inflammatory, and antiproliferative effects of the drug. In fact, there may be “pleiotropic” consequences of platelet inhibition by low-dose aspirin, resulting from reduced release of platelet-active prostanoids, inflammatory cytokines, and growth factors.1 The recent reports18, 19 of long-term reduction in the risk of colorectal (and other) cancer and death associated with low-dose aspirin in RCTs using as low as 75 mg daily are consistent with this hypothesis.20
Assessing the benefit/risk profile of aspirin requires an estimation of the absolute risk of the individual patient for atherothrombotic and hemorrhagic complications. In individuals at low risk for vascular occlusion (e.g., <1% per year), a very small absolute benefit may be offset by exposure of very large numbers of healthy subjects to undue serious bleeding complications (see Balance of Benefits and Risks). As the risk of experiencing a major vascular event increases, so does the absolute benefit of antiplatelet prophylaxis with aspirin (FIGURE 111.3), and, above a certain threshold, benefit clearly outweighs risk of bleeding (Table 111.1).2, 5
Table 111.1 Benefit/risk ratio of antiplatelet prophylaxis with aspirin in different settings
Number of Patients in Whom a Major Vascular Event is Avoided per 1,000/y
Number of Patients in Whom a Major GI Bleeding Event is Caused per 1,000/y
Men and women at low cardiovascular risk
1-2
1-2
1
Essential hypertension
1-2
1-2
1
Chronic stable angina
10
1-2
5-10
Prior stroke or TIA
10
1-2
5-10
Prior myocardial infarction
15
1-2
7.5-15
Unstable angina
50
1-2
25-50
a Benefits are calculated from randomized trial data reviewed in Refs. 2, and 5 and depicted in FIGURE 111.3 .
b Risks of upper GI bleeding are estimated from a background rate of 1 event per 1,000 per year in the general population of nonusers and a RR of 2.0 to 3.0 associated with aspirin prophylaxis. Such an estimate assumes comparability of other risk factors for upper GI bleeding, such as age and concomitant use of NSAIDs, and may actually underestimate the absolute risk in an elderly population exposed to “primary” prevention. The absolute excess of major extracranial bleeding complications in the “primary” prevention trials reviewed in Ref. 39 ranged between 0.2 and 2.0 per 1,000 patient-years.
Modified from Patrono C, Baigent C, Hirsh J, et al. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008;133:199S-233S.
The efficacy and safety of aspirin has been evaluated in numerous placebo-controlled RCTs of patients at variable risk of atherothrombotic complications, from asymptomatic low-risk individuals to patients presenting with an acute myocardial infarction or an acute ischemic stroke2, 5; trials have extended from as short as a few weeks’ duration to as long as 10 years. In the Second International Study of Infarct Survival,21 a single tablet of aspirin (162.5 mg) started within 24 hours of the onset of symptoms of a suspected myocardial infarction and continued daily for 5 weeks produced highly significant reductions in the risk of vascular mortality (by 23%), nonfatal reinfarction (by 49%), and nonfatal stroke (by 46%). There was no increase in hemorrhagic stroke or GI bleeding in the aspirin-treated patients and only a small increase in minor bleeding.21 Treatment of 1,000 patients with suspected acute myocardial infarction with aspirin for 5 weeks will result in approximately 40 patients in whom a vascular event is prevented,22 with a proportional odds reduction of 30%. Two separate trials with a similar protocol, the International Stroke Trial23 and the Chinese Acute Stroke Trial,24 tested the efficacy and safety of early aspirin use in acute ischemic stroke. Approximately 40,000 patients were randomized within 48 hours of the onset of symptoms to 2 to 4 weeks of daily aspirin therapy (300 and 160 mg, respectively) or placebo. An overview of the results of both trials suggests an absolute benefit of nine fewer deaths or nonfatal strokes per 1,000 patients in the 1st month of aspirin therapy.22 The proportional odds reduction in fatal or nonfatal vascular events was only 10% in this setting. Although the background risk of hemorrhagic stroke was threefold higher in the Chinese Acute Stroke Trial than in the International Stroke Trial, the absolute increase in this risk associated with early use of aspirin was similar in the two studies (excess 2 per 1,000 patients).23, 24
Balance of Benefits and Risks
Long-term aspirin therapy confers conclusive net benefit on the risk of subsequent myocardial infarction, stroke, or vascular death among subjects at high risk of vascular complications. These include patients with chronic stable angina,25 patients with prior myocardial infarction,22 patients with unstable angina,26, 27, 28, 29 and patients with transient ischemic attack (TIA) or minor stroke30, 31, 32, 33, 34, 35 as well as other high-risk categories.22 The proportional effects of long-term aspirin therapy on vascular events in these different clinical settings showed substantial heterogeneity, ranging from no statistically significant benefits in patients with peripheral vascular disease36 to approximately 50% risk reduction in patients with unstable angina.22 In terms of absolute benefit, these protective effects of aspirin translate into avoidance of a major vascular event in 50 per 1,000 patients with unstable angina treated for 6 months and in 36 per 1,000 patients with prior myocardial infarction, stroke, or TIA treated for approximately 30 months.22
For patients with different manifestations of ischemic heart or brain disease, a consensus exists in defining a rather narrow range of recommended daily doses (i.e., 75 to 160 mg) for the prevention of atherothrombotic complications.37, 38 This is supported by separate trial data in patients randomized to treatment with low-dose aspirin or placebo as well as by an overview of all antiplatelet trials showing no obvious dose dependence, from indirect comparisons, for the protective effects of aspirin.22 There is no convincing evidence that the dose requirement for the antithrombotic effect of aspirin varies in different clinical settings.2
Among most high-risk patient groups, the expected number avoiding a serious vascular event by using aspirin substantially exceeds the number experiencing a major bleed (Table 111.1). However, it is unclear whether aspirin might benefit people who, although asymptomatic, are at low to intermediate risk of serious vascular events. The question of whether aspirin is effective and safe for the primary prevention of vascular events has been addressed in a meta-analysis of six randomized trials.39 It is interesting to compare the effects of low-dose aspirin in primary prevention with the well-known benefits in secondary prevention.39 In the six primary prevention trials among 95,000 low-risk individuals, with a mean follow-up of 6.9 years, aspirin allocation yielded a 12% RR reduction in serious vascular events, from an annual rate of 0.57% to 0.51% (FIGURE 111.4). This effect was mainly due to a reduction in nonfatal myocardial infarction, from 0.23% to 0.18% per year. The net effect on stroke was not significant (from 0.21% to 0.20% per year), reflecting a small reduction in presumed ischemic stroke and counterbalancing effects on hemorrhagic stroke and other stroke.39 There was no significant reduction in vascular mortality (from 0.19% to 0.19% per year). Aspirin allocation increased GI (or other extracranial) bleeds by about half, from 0.07% to 0.1% per year.39
Only gold members can continue reading. Log In or Register to continue
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



