Antifolates
The antifolates were introduced as antileukemic drugs in 1948; in landmark experiments treating children with acute lymphocytic leukemia (ALL), Sidney Farber produced the first evidence that chemotherapy with a folate analogue, aminopterin, could lead to complete remissions (1). Methotrexate subsequently became the standard antifolate in treatment of ALL. It has since gained an important role in regimens for lymphomas, and choriocarcinoma, and as an immunosuppressive following allogeneic bone marrow transplantation. It is also a standard agent for treating rheumatoid arthritis, Wegener’s granulomatosis, and other inflammatory/autoimmune diseases. Pemetrexed (Alimta), a closely related structure but with a different site of action, is widely used for non-small cell lung cancer, mesothelioma, and ovarian cancer. Pralatrexate, the newest antifolate similar in action to methotrexate, is highly active against peripheral T-cell lymphoma and cutaneous T-cell lymphoma.
The structures of antifolates are shown in Figure 2-1. The analogues closely resemble naturally occurring folates, but contain substitutions in the basic pteridine ring system, as in pralatrexate and pemetrexed, which enhance binding and transport. The key addition of the amino group on the C-4 position of the pteridine ring, as found in methotrexate, enhances inhibition of dihydrofolate reductase. Changes in the bridge system connecting the unsaturated rings to para-aminobenzoyl glutamate (PABG) enhance the active uptake and polyglutamation of pemetrexed and pralatrexate. Because of their strong electronegative charge at physiologic pH, the parent antifolates, like physiologic folates, require active transport into cells via the reduced folate carrier (2). In selected cells, such as choriocarcinomas, a second carrier, the folate binding protein, mediates folate and methotrexate transport, and becomes the preferred transporter. Pemetrexed is also transported by a third carrier, the proton-coupled folate carrier, which may be responsible for its unique activity against epithelial cancer and mesothelioma (3). Inside the cell, the analogues, like the physiologic folates, are converted at their PABG terminus to highly charged, long-chain polyglutamates. These polyglutamate metabolites are retained preferentially within cells and inhibit, with increased affinity, a number of folate-dependent enzymes critical for both thymidine and purine biosynthesis (Figure 2-2). Through their inhibition of dihydrofolate reductase, methotrexate and pralatrexate deplete intracellular folates, leading to a block in both purine and pyrimidine biosynthesis. The primary action of pemetrexed is its inhibition of another folate-dependent enzyme, thymidylate synthase.
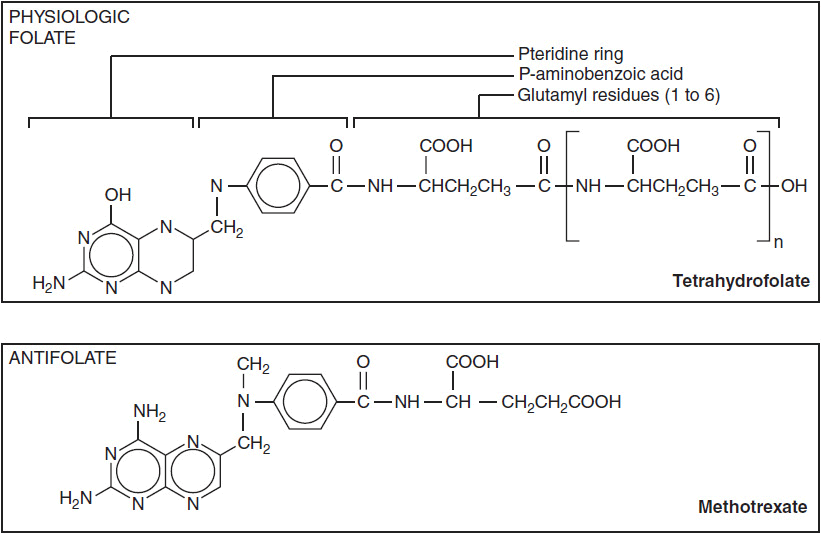
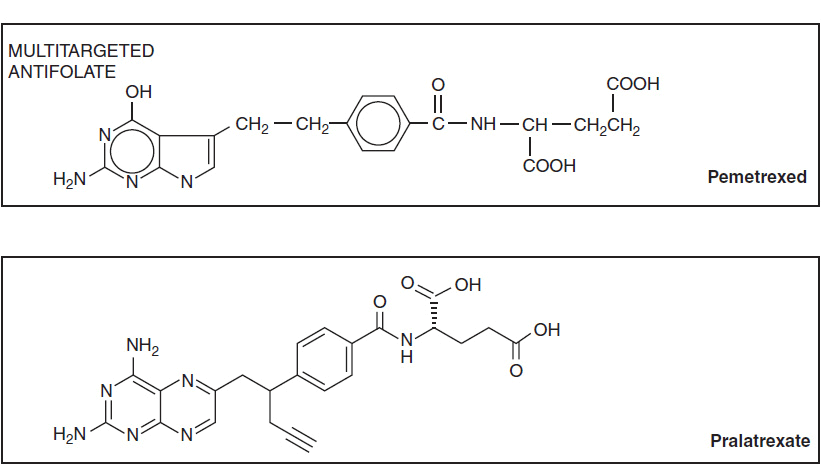
FIGURE 2-1 Molecular structure of folic acid, methotrexate, pemetrexed, and pralatrexate.
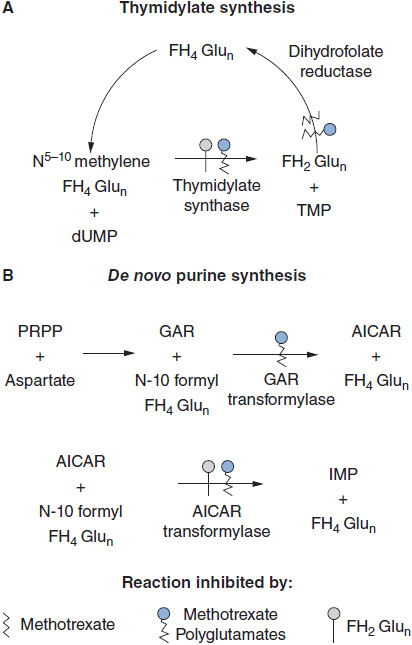
FIGURE 2-2 Multiple sites of inhibitory action of methotrexate, its polyglutamate metabolites, and dihydrofolate polyglutamates, the substrate that accumulates when dihydrofolate reductase is inhibited. AICAR: aminoimidazole carboxamide; TMP: thymidine monophosphate; dUMP: deoxyuridine monophosphate; FH2Glun: dihydrofolate polyglutamate; FH4Glun: tetrahydrofolate polyglutamate; GAR: glycinamide ribonucleotide; IMP: inosine monophosphate; PRPP: 5-phosphoribosyl-1-pyrophosphate. Pralatrexate acts in a very similar fashion, while the polyglutamates of pemetrexed primarily act as direct inhibitors of TS.
PHARMACOLOGIC CONSIDERATIONS
Methotrexate and Pralatrexate antifolates kill cells through depletion of thymidylate and purine precursors of DNA, and are thus most effective against rapidly growing tumors, such as leukemias and lymphomas. Cell kill by methotrexate depends on both drug concentration and duration of exposure. Its threshold concentration for drug toxicity for normal cells lies in the range of 10 nM. Methotrexate-resistant cells arise through loss of the reduced folate transporter, loss of the ability to polyglutamate antifolates, or amplification of the gene coding for dihydrofolate reductase, all of which have been demonstrated in ALL cells in association with relapse (4, 5). High levels of TS expression are associated with poor response to pemetrexed (4), while resistance to pralatrexate likely parallels that of methotrexate but is incompletely understood.
Inherited genetic variants modify folate metabolism (6, 7). The methylene tetrahydrofolate reductase variant C677T increases the intracellular level of 5–10 methylene tetrahydrofolic acid, the substrate for TS, and is associated with an increased rate of relapse in childhood ALL (7). Attempts to relate response and toxicity to variations in transporter and polyglutamate synthase genes have been inconclusive.
Pemetrexed and pralatrexate are more avidly transported and converted to a polyglutamate than is methotrexate (8). Because of its mild and predictable toxicities, pemetrexed has become a preferred agent for first- or second-line treatment of adenocarcinoma of the lung. Unlike methotrexate, pemetrexed and pralatrexate are given with folic acid (0.4–1.0 mg/day, beginning 1 week prior to treatment, and continuing throughout therapy) and with vitamin B12 (1 μg on day 1 i.m. and thereafter with each cycle of therapy), as these vitamins ameliorate toxicity to bone marrow (
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


