The folate-dependent enzymes represent attractive targets for antitumor chemotherapy because of their critical role in the synthesis of the nucleotide precursors of DNA (Fig. 8-1). In 1948, Farber et al.1 were the first to show that aminopterin, a 4-amino analog of folic acid, could inhibit the proliferation of leukemic cells and produce remissions in acute leukemia cases. Their findings ushered in the era of antimetabolite chemotherapy and generated great interest in the antifolate class of agents. Since then, antifolate compounds have become extremely valuable in the treatment of hematologic and nonhematologic malignancies as well as nonneoplastic disorders, including rheumatoid arthritis,2 psoriasis,3 and bacterial, fungal, and parasitic infections.4 High-dose regimens have further expanded the role of methotrexate (MTX) and have become the mainstay of therapy for primary central nervous system lymphomas and for prevention of meningeal leukemia,5 where it has replaced cranial irradiation in children with acute lymphoblastic leukemia (ALL). At this time, the antifolates are one of the best understood and most versatile of all the cancer chemotherapeutic drug classes. The key features of MTX, the most commonly used antifolate, are presented in Table 8-1.
Mechanism of Action
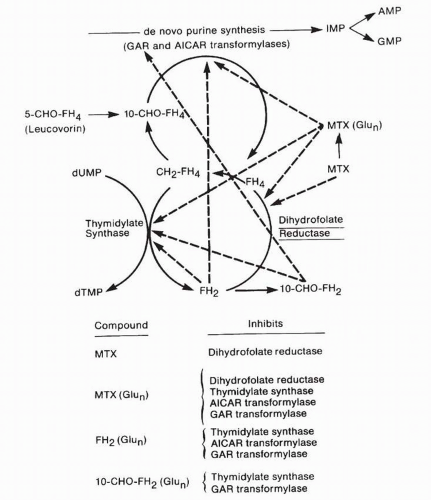
Substitution of an amino group for the hydroxyl at the 4-N position of the pteridine ring critically changes the structure of folate compounds, leading to their enzyme inhibitory and antitumor activities. This change transforms the molecule from a substrate to a tight-binding inhibitor of dihydrofolate reductase (DHFR), a key enzyme in intracellular folate homeostasis. The critical importance of DHFR stems from the fact that folic acid compounds are active as coenzymes only in their fully reduced, tetrahydrofolate, form. Two specific tetrahydrofolates play essential roles as one-carbon carriers in the synthesis of DNA precursors. The cofactor 10-formyltetrahydrofolate provides its one-carbon group for the de novo synthesis of purines in reactions mediated by glycineamide ribonucleotide (GAR) transformylase and aminoimidazole carboxamide ribonucleotide (AICAR) transformylase. A second cofactor, 5,10-methylenetetrahydrofolate (CH2-FH4), donates its one-carbon group to the reductive methylation reaction that converts deoxyuridylate (dUMP) to thymidylate (TMP) (Fig. 8-2). In addition to contributing a one-carbon group, CH2FH4 is oxidized to dihydrofolate (FH2), which must then be reduced to tetrahydrofolate by the enzyme DHFR to enable it to rejoin the pool of active reduced-folate cofactors. In actively proliferating tumor cells, inhibition of DHFR by MTX or other 2,4-diamino antifolates leads to an accumulation of folates in the inactive FH2 form, with variable depletion of reduced folates.6, 7, 8, 9, 10, 11, 12 Folate depletion, however, does not fully account for the metabolic inhibition associated with antifolate treatment because the critical reduced-folate pools may be relatively preserved even in the presence of cytotoxic concentrations of MTX. Additional factors may contribute to MTX-associated cytotoxicity, including metabolism of the parent compound to polyglutamated derivatives and the accumulation of dihydrofolate polyglutamates (PGs) as a consequence of DHFR inhibition.6,7,13, 14, 15 MTX and dihydrofolate PGs represent potent direct inhibitors of the folate-dependent enzymes of thymidylate and purine biosynthesis.16, 17, 18, 19 Thus, inhibition of DNA biosynthesis by 2,4-diamino folates is a multifactorial process consisting of both partial depletion of reduced-folate substrates and direct inhibition of folate-dependent enzymes. The relative roles of each of these mechanisms in determining antifolate-associated metabolic inhibition may depend on factors specific to various cancer cell lines and tumors.
Chemical Structure
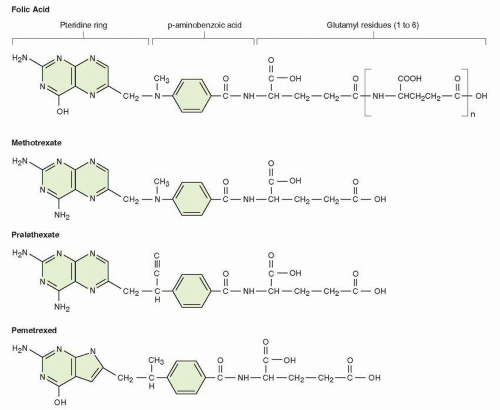
The physiological folate structure, as shown in Figure 8-1, consists of a pteridine ring, which connects via an N-10 bridge to benzoic acid and thence to a terminal glutamic acid. Various heterocyclic compounds with the 2,4-diamino configuration may bind to DHFR and have antifolate activity in microbial organisms and include pyrimidine analogs such as pyrimethamine and trimethoprim18, 19, 20, 21, 22, 23, 24, 25; the antimicrobial compounds are in general weak inhibitors of the mammalian DHFR. The antifolates with DHFR inhibitory activity conserve the inhibitory 2,4-diamino configuration but have the added structural features of folates; these compounds include classical pteroyl glutamates such as aminopterin and MTX2; compounds with an altered pteridine ring, such as those with replacement of the 5- or 8-N position, or both, with a carbon atom (the quinazolines) lipid-soluble derivatives lacking a terminal glutamate (trimetrexate and piritrexim22,23); or folate-like compounds with alterations in the N-10 bridge (pralatrexate24). Each of these changes alters the pharmacological properties (transport, enzyme binding, and polyglutamation) of the folate inhibitory molecule, as described below. Investigators have designed antifolate analogs directed at targets other than DHFR, such as those folate-dependent enzymes required for the de novo synthesis of purines and thymidylate synthase (TS). Potent TS inhibitors, which utilize physiological pathways for transport and polyglutamation, include raltitrexed (ZD1694, Tomudex) and pemetrexed (LY231514, Alimta).25, 26, 27, 28 The antifolates conserve the 2-amino, 4-hydroxy configuration of physiological folates and are weaker inhibitors of DHFR. Pemetrexed is approved for marketing in the United States and elsewhere, based on its activity in mesothelioma and adenocarcinoma of the lung. MTX is considered first in this chapter.
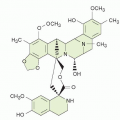
FIGURE 8-1 Structure of folic acid PG, showing its component subgroups (pteroic acid, para-amino benzoic acid, glutamate). Also shown are the analogues methotrexate, pralatrexate, and pemetrexed, which preserve most of the essential features of folates and are readily converted to PGs within cells. MTX and pralatrexate contain the critical 2, 4 diamino configuration that inhibits DHFR. Pemetrexed inhibits TS and GARTF (see text).
Cellular Pharmacology and Mechanisms of Resistance
In this section, the sequence of events that leads to the cytotoxic action of MTX is considered, beginning with drug movement across the cell membrane, followed by its intracellular metabolism to the PG derivatives, binding to DHFR and other folate-dependent enzymes, effects on intracellular folates, and, finally, inhibition of DNA synthesis.
Transmembrane Transport
Folate influx into mammalian cells proceeds via three distinct transport systems: (a) the reduced-folate carrier (RFC) system, (b) the folate receptor (FR) system (Table 8-2),29, 30, 31, 32 and (c) a pH-sensitive transporter first identified in the intestinal epithelium and found in most tissues and in selected tumor cells33 (see Table 8-2). The proliferative or kinetic state of tumor cells, as well as the temperature and pH of the extracellular environment, influences the rate of folate and MTX transport. In general, rapidly dividing cells have a greater rate of MTX uptake and a lower rate of drug efflux than cells that are either in the stationary phase or that are slowly growing.34 The RFC system, with its large transport capacity, transports folic acid inefficiently (Kt [transport coefficient] = 200 μmol/L) and is a primary transport mechanism of the reduced folates and antifolates like MTX (Kt = 0.7 to 6.0 μmol/L), at pharmacologic drug concentrations.35, 36, 37 The RFC system also transports the naturally occurring reduced folates, including the rescue agent 5-formyltetrahydrofolate (leucovorin).30,31,38 The RFC gene resides on the long arm of chromosome 21 and encodes a protein with predicted molecular size of 58 to 68 kD.39,40 Mutations in the RFC, affecting glutamic acid residue 45 and other sites, have been associated with MTX resistance.41, 42, 43 The proton-coupled transporter may become the predominant folate carrier under relatively acidic conditions, as may be expected in poorly perfused areas of solid tumor masses.33,44
TABLE 8.1Key features of methotrexate sodium (MTX)
Mechanism of action
Inhibition of DHFR leads to partial depletion of reduced folates PGs of MTX and dihydrofolate inhibit purine and thymidylate biosynthesis
Metabolism
Converted to PGs in normal and malignant tissues. 7-Hydoxylation in liver
Pharmacokinetics
t½α = 2-3 h; t½β = 8-10 h
Elimination
Primarily as intact drug in urine
Drug interactions
Toxicity to normal tissues rescued by leucovorin calcium L-Asparaginase blocks toxicity and antitumor activity Pretreatment with MTX increases 5-fluorouracil and cytosine arabinoside nucleotide formation Nonsteroidal anti-inflammatory agents decrease renal clearance and increase toxicity
Reduce dose in proportion to creatinine clearance Do not administer high-dose MTX to patients with abnormal renal function Monitor plasma concentrations of drug, hydrate patients during high-dose therapy (see Tables 8-3 and 8-4)
t½, half-life
A second folate transport mechanism consists of isoforms of the FR, which mediates the internalization of folates via a high-affinity membrane-bound 38-kD glycoprotein. The FR gene family encodes three homologous glycoproteins that share a similar folate-binding site. The α and β FRs are anchored to the plasma membrane by a carboxyl-terminal glycosylphosphatidylinositol tail and transport the reduced folates and MTX at a lower capacity than the RFC system. The function of the FR-γ is unknown. The FRs are expressed in normal tissues and, at high levels, on the surface of some epithelial tumors such as ovarian cancer.45,46
Source: Mauritz R, Peters GJ, Kathmann I, et al. Dynamics of antifolate transport via the reduced folate carrier and the membrane folate receptor in murine leukemia cells in vitro and in vivo. Cancer Chemother Pharmacol 2008;62:937-948.
FIGURE 8-2 Sites of action of methotrexate (MTX), its polyglutamated metabolites (MTX[(Glun]), and folate byproducts of the inhibition of DHFR, including dihydrofolate (FH2) and 10-formyldihydrofolate (10-CHO-FH2). Also shown are 5,10-methylenetetrahydrofolate (CH2-FH4), the folate cofactor required for thymidylate synthesis and 10-formyltetrahydrofolate (10-CHO-FH4), the required intermediate in the synthesis of purine precursors. AICAR; aminoimidazole carboxamide ribonucleotide; AMP, adenosine monophosphate; dUMP, deoxyuridylate; dTMP, thymidylate; GAR; glycineamide ribonucleotide; GMP, guanosine monophosphate; IMP, inosine monophosphate. (Dashed lines indicate inhibition.) (From DeVita VT, Hellman S, Rosenberg SA, eds. Cancer: Principles and Practice of Oncology. Philadelphia: JB Lippincott, 1989:349-397.)
The FR system has a higher affinity for folic acid and the reduced folates (1 nM) than for MTX (5 to 10 nM). In addition, MTX PGs demonstrate a 75-fold increased affinity for FR compared with the monoglutamate form of MTX.47 The FR is strongly expressed on ovarian cancer cells as the CA-125 antigen and is now the target of experimental monoclonal antibody therapy.48 Variation in exogenous folate concentrations and normal physiologic conditions, such as pregnancy, can alter the tissue expression of FR. Intracellular levels of homocysteine, which increase under folate-deficient conditions, are a critical modulator of the translational up-regulation of FRs.49 Under conditions of relative folate deficiency, elevated levels of homocysteine stimulate the interaction between heterogeneous nuclear ribonucleoprotein E1 and an 18-base-pair region in the 5′-untranslated region of the FR mRNA resulting in increased translational efficiency and therefore elevated cellular levels of FR protein. This mechanism of transcriptional regulation through protein binding to its message is operative in DHFR and TS protein synthesis as well.
The FR isoforms (α, β, γ) are independently expressed in mammalian cells and normal human tissues.50 FR-α is expressed on the apical and luminal surfaces of placenta, chorioid plexus, renal tubules, and alveolar cells and in human epithelial neoplasms (ovarian cancer, papillary serous endometrial cancer, renal cell cancers, and non-small cell lung cancers).51 In nasopharyngeal KB carcinoma cells, FR-α is up-regulated by folate depletion and down-regulated in folate-replete medium.29,36 Elwood et al.52 and others53,54 have shown that FR-α expression is regulated at a molecular level by promoters upstream from exons 1 and 4 and by differential messenger RNA (mRNA) splicing of 5′ exons. FR-β is expressed in human placenta and nonepithelial tumors. FR-γ, found in hematopoietic and lymphatic cells and tissues, lacks a glycosylphosphatidylinositol membrane anchor and is secreted. Although human FR-α, FR-β, and FR-γ share 70% amino acid sequence homology, they differ in binding affinities for stereoisomers of folates.55
The precise mechanism of FR-mediated folate uptake remains controversial56; two separate pathways for FR-mediated folate uptake have been reported: (a) the classic receptor-mediated internalization of the ligand-receptor complex through clathrin-coated pits with subsequent formation of secondary lysosomes, and (b) a mechanism of small molecule uptake, termed potocytosis,57, 58, 59 in which receptor complexes accumulate within distinct subdomains of the plasma membrane known as caveolae that internalize to form intracellular vesicles.60 Once internalization has occurred, acidification within the vesicle causes the folate-receptor complex to dissociate and translocate across the cell membrane. Although questions remain as to the relative importance of the FR and RFC transport systems in the uptake of antifolates during chemotherapy, studies suggest that the RFC system is the more relevant transporter of MTX in most mammalian cells and tumors.61,62
The third transporter for folates and analogues, the low-pH, proton-coupled transporter, was first identified in intestinal cells, but is found in many normal tissues as well as tumors, and mediates folate transport into the central nervous system.63,64 It utilizes an inwardly directed H+ gradient to move folates and analogues into cells and is lacking in an inherited folate malabsorption disorder.33,64 It efficiently transports pemetrexed (Km = 0.2 to 0.8 μM) but has a lower affinity for MTX and reduced folates.
Role of Transporters in Resistance to MTX
Both in vitro and in vivo experimental systems and limited clinical studies have identified decreased transport as a common mechanism of intrinsic or acquired resistance to MTX. A number of MTX-resistant cell lines with functional defects in the RFC have now been described.65, 66, 67, 68 An MTX-resistant human lymphoblastic CCRF-CEM/MTX cell line maintained in physiologic (micromolar) concentrations of folate (2 nmol/L) lacked the RFC protein and, for this reason, were resistant to MTX.69 These cells retained the FR, however, and used this transport process to maintain growth even in nanomolar concentrations of folic acid.
A mutated murine RFC (RFC1) with increased affinity for folic acid and decreased affinity for MTX contained an amino acid substitution at glutamic acid residue 45. This region of the RFC is also a cluster site for mutations that occur when cells are placed under selective pressure with antifolates that use RFC1 as the major route of entry into mammalian cells.70,71 However, none of 121 samples of ALL cells from drug-resistant patients contained a mutation of glutamic acid residue 45.72
Other studies provide suggestive but inconclusive evidence for a role of transport deficiency in MTX resistance in both ALL and in solid tumors.73, 74, 75, 76, 77, 78 A sensitive competitive displacement assay using the fluorescent analog of MTX73 revealed that blast cells from two of four patients in relapse after initial treatment with MTX-based combination chemotherapy demonstrated defective MTX uptake. In two studies of patients with newly diagnosed ALL, low RFC expression at diagnosis correlated with a significantly reduced event-free survival.74,75 In osteosarcoma treatment, 17 of 26 posttreatment tumor samples (65%) derived from patients with high-grade tumors and with poor response to chemotherapy had decreased RFC expression.76 Poor response to MTX-based chemotherapy was also observed in tumor samples with low levels of RFC at diagnosis.77 Thus, impaired transport of MTX may be a common mechanism of intrinsic resistance in osteosarcoma.
Some newer antifolate analogues have improved transport properties. The lipid-soluble nonglutamated antifolates such as trimetrexate and piritrexim, as well as the glutamyl esters of MTX, do not require active cellular transport and demonstrate activity against transport-resistant mutants79,80 but lack polyglutamation and have minimal activity as antitumor agents. Pralatrexate, the newest addition to the field and now approved for cutaneous T-cell lymphomas, is one of several 10-deaza-aminopterins with a 10-fold or greater efficiency than MTX as a substrate for the reduced folate transporter.81,82 In contrast to MTX (which has a relatively poor affinity for FR), other antifolate inhibitors (CB3717, raltitrexed, DDATHF, [LY231514], and BW1843U89) rely on the FR for transport.83,84 Because pemetrexed is efficiently transported by both FR and proton-coupled folate transport systems, it may be less susceptible to the emergence of clinical resistance resulting from alterations in RFC.85,86
Low folate conditions may increase the toxicity of antifolates as a consequence of the up-regulations of FR in normal tissues in response to folate starvation.87 Folate supplementation of patients receiving either pemetrexed or pralatrexate reduces the incidence of severe myelosuppression related to these drugs.88
MTX Efflux Mechanisms
Early studies suggested the presence of active efflux mechanisms for MTX and the folates.89, 90, 91 Subsequent experiments have revealed that the multidrug resistance-associated protein family of ATP-binding cassette transporters, particularly MRP-1, MRP-2, and MRP-3 and the breast cancer resistance protein (BCRP), actively extrude MTX in both normal and tumor cells, affecting both drug pharmacokinetics and resistance.92, 93, 94, 95 MRP-2 and BRCP or ABCG2 excrete MTX from liver cells into the bile and efflux drug into the intestinal lumen and urine in mice. Knockout-of BCRP produces increased MTX levels in the systemic circulation in mice as a consequence of decreased biliary and urinary excretion.
Experiments in mice suggest that efflux alterations may contribute to MTX resistance. Overexpression of MRP-1 was associated with MTX resistance in murine tumor experiments, and inhibitors of MRP-1 (probenecid) reversed antifolate resistance.96 Interestingly, others found that loss of MRP-1 expression may expand intracellular folate pools and result in antifolate resistance.97 BCRP is associated with increased cellular efflux of both MTX and PGs with two or three glutamic acid residues, and its overexpression or mutations may lead to MTX resistance.98
Intracellular Transformation
Naturally occurring folates exist within cells in a polyglutamated form (Fig. 8-1). The polyglutamation of folate substrates is directed by folylpolyglutamyl synthetase (FPGS), an enzyme that adds up to eight glutamyl groups in γpeptide linkage. This reaction serves several purposes for folates and antifolates: (a) it facilitates the accumulation of intracellular folates in a selectively retained form, in vast excess of a monoglutamate pool that is freely transportable into and out of cells, (b) it allows selective intracellular retention of these relatively large anionic molecules and thus prolongs intracellular half-life, under conditions of reduced folate availability, and (c) it enhances folate cofactor affinity for folate-dependent enzymes. The MTX PGs are slightly more potent inhibitors of DHFR but significantly more potent antagonists of TS, AICAR transformylase, and GAR transformylase as compared to the parent, MTX-Glu1.13,14 MTX and the other glutamyl-terminal analogs also undergo polyglutamation in normal liver cells and bone marrow myeloid precursors,99,100 likely enhancing their toxicity. Polyglutamation occurs in most tumors to varying degrees and likely determines antitumor response.100, 101, 102, 103
The efficiency of the polyglutamation reaction for various folate substrates and antifolates varies considerably. Pralatrexate is 10-fold more avidly polyglutamated than MTX.88 The polyglutamation of MTX occurs progressively over several hours of exposure; after 24 hours, 80% or more of intracellular drug exists in the PG form.101,104, 105, 106 Human liver retains MTX PGs for several months after drug administration.107 Thus, selective retention and depot formation in excess of free monoglutamate, as seen with physiologic folates, characterize MTX PGs as well.
FPGS is a 62-kD magnesium-, adenosine triphosphate-, and potassium-dependent protein.108, 109, 110, 111 The FPGS gene is located on chromosome region 9q34.11 and produces two proteins, the major one having 537-amino acids and a second with 579 amino acids found in mitochondria. The most avid substrate for this enzyme is dihydrofolate (Km [binding affinity] = 2 μmol/L), with the following folates and analogues in descending order of affinity: tetrahydrofolate (Km = 6 μmol/L) > 10-formyltetrahydrofolate or 5-methyltetrahydrofolate > aminopterin > leucovorin > MTX. Because of the relatively slow rate of formation of MTX PGs compared with the naturally occurring folate PGs, reductions in FPGS activity or cellular glutamate levels may have little effect on folate PG pools but may critically reduce MTX PG formation and decrease cytotoxicity of MTX and likely other antifolates.
The intracellular content of PG derivatives represents a balance between the activity of two different enzymes: FPGS, which synthesizes PGs, and γ-glutamyl hydrolase (GGH, conjugase),112 a γ-glutamyl-specific peptidase that removes terminal glutamyl groups and returns MTX PGs to their monoglutamate form.113
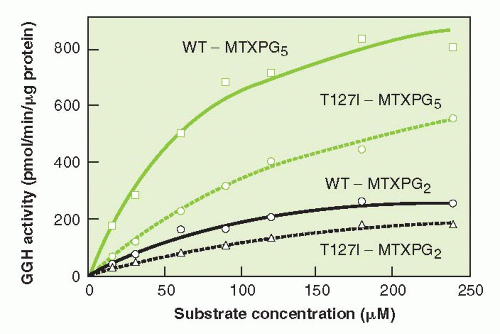
While the functional importance of GGH in determining response to MTX is not known, the St. Jude group has found a substrate specific polymorphism (C452T) of the enzyme, present in 10% of Caucasian patients, that reduces catalytic activity and allows greater accumulation of MTX PGs in leukemic cells (Fig. 8-3).114 Yao et al.115 isolated and cloned the complementary DNA (cDNA) for GGH, which codes for an enzyme of 318 amino acids and has a molecular weight of 36 kD. Although it may be expected that overexpression of hydrolase may result in MTX resistance, particularly with brief drug exposures, such was not the case in several human cell line models in which hydrolase was overexpressed.116
MTX PGs exist essentially only within cells and enter or exit cells sparingly in vitro.102,117 The diglutamate form has an uptake velocity of one-fifteenth that of MTX,118 whereas higher glutamates have even slower transport rates.102,117 Thus, MTX PGs are selectively retained in preference to parent drug as extracellular levels of MTX fall.
Cellular activity of FPGS correlates directly with the rate of cell growth103,119 and inversely with the level of intracellular folates.119,120 Enhancement of cell proliferation with growth factors such as insulin, dexamethasone, tocopherol, and estrogen in hormone-responsive cells increases polyglutamation, whereas deprivation of essential amino acids121 results in inhibition of polyglutamation. MTX and L-asparaginase are frequently used in combination for the treatment of acute leukemia. Conversion of MTX to PG forms can be markedly inhibited by pretreatment with L-asparaginase, presumably through amino acid deprivation with resultant growth arrest.122 Increasing intracellular folate pools through exposure of cells to high concentrations of leucovorin or 5-methyltetrahydrofolate results in a decrease in MTX polyglutamation.120 Conversely, the process is enhanced in human hepatoma cells either by incubating cells with MTX in folate-free medium or by first depleting the intracellular folates by “permeabilizing” cell membranes in a folate-free environment.119
FIGURE 8-3 Catalytic activity of T127I variant gamma-glutamylhydrolase (GGH) on MTX PG substrates, as compared to wild type (WT) enzyme. Enzymes were incubated with MTX PGs with chain length of 2 and 5 glutamyl groups. A 2.7 fold increase in Km was observed for the variant enzyme. (From Cheng Q, et. al. A substrate specific functional polymorphism of human gamma-glutamyl hydrolase alters catalytic activity and methotrexate polyglutamate accumulation in acute lymphoblastic leukemia cells. Pharmacogenetics 2004;14:557-567.)
An important factor in the selective nature of MTX cytotoxicity may derive from modest PG formation in normal tissues relative to that in malignant tissues. Although little metabolism to PGs is observed in normal murine intestinal cells in vivo, most murine leukemias and Ehrlich ascites tumor cells efficiently convert MTX to higher PG forms in tumor-bearing animals.37,123 Additionally, normal human and murine myeloid progenitor cells form relatively small amounts of MTX PGs compared with leukemic cells.99,100
In addition to increasing its retention within cells, polyglutamation of MTX enhances its inhibitory effects on specific folate-dependent enzymes. The pentaglutamates have a slower dissociation rate from DHFR than does MTX124 and a markedly enhanced inhibitory potency for TS (Ki = 50 nmol/L), AICAR transformylase (Ki = 57 nmol/L), and, to a lesser extent, GAR transformylase (Ki = 2 μmol/L) in the presence of monoglutamated folate substrates.12 The well-described incomplete depletion of physiologic folate cofactors by MTX suggests that direct enzymatic inhibition by MTX PGs may contribute to MTX cytotoxicity. These effects may also explain the competitive nature of leucovorin rescue and the relatively selective rescue of normal versus malignant tissues, in that rescue may depend on the ability of leucovorin and its derived tetrahydrofolates to compete with MTX PGs at sites other than DHFR.
The ability of antifolate analogs to undergo polyglutamation is one of several properties that influence cytotoxic potency. Aminopterin is a better substrate for FPGS than is MTX and is a more potent cytotoxic agent. A fluorinated MTX analog, PT430, is a weak substrate for FPGS and has little cytotoxic activity.125 As mentioned previously pemetrexed and particularly pralatrexate are more efficient substrates than MTX. The ability to generate PGs has been correlated with sensitivity to MTX and to other antifolate agents that undergo polyglutamation, including pemetrexed and raltitrexed, and is often defective in drug-resistant human and murine tumor cell lines.126, 127, 128, 129, 130
Although defective polyglutamation may coexist with other metabolic alterations, examples of pure polyglutamation defects have been described in human leukemia cell lines (CCRF-CEM)131,132 and in human squamous cancer cell lines derived from head and neck tumors and have been implicated as the specific cause of MTX resistance in human B- and T-cell types of ALL.132, 133, 134, 135, 136, 137, 138, 139, 140, 141
A decrease in MTX PGs was found in small cell carcinomas resistant in vitro after clinical treatment with MTX.135 In ALL, the balance of FPGS and GGH activities before treatment predicted PG formation and therapeutic outcome in a small prospective trial.136 Examples of defective splicing of the folylpolyglutamate mRNA were identified in cell lines selected for MTX resistance, exon 12 skipping was found in bone marrow samples from two ALL patients at diagnosis and one at relapse after MTX therapy.132 Another study reported a decreased binding affinity (higher Km) for MTX as a substrate for FPGS in enzyme from blast cells of patients with acute myelogenous leukemia (AML) as opposed to ALL cells. This difference in affinity resulted in a predominance of MTX-Glu1 species in AML cells, and MTX Glu3-5 in ALL cells.
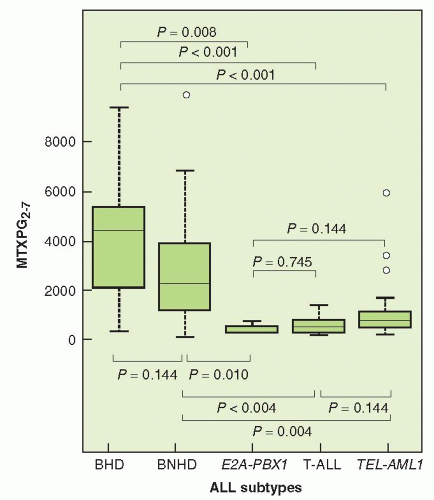
Leukemic cells differ greatly in their expression of FPGS (Fig. 8-4). Hyperdiploid status in childhood ALL is a good prognostic feature; cells with hyperdiploid chromosomes show higher levels of synthesis of cytotoxic MTX PGs than cells of diploid, T-cell, TEL-AML1 or E2APBX1 lymphoblasts.114 Investigators have found a higher concentration of MTX long-chain PGs in B than in T lymphoblasts and an increased level of expression of FPGS mRNA in B-lineage cells.137,138 These findings suggest that the higher response rates observed in patients with B-cell ALL may result from increased levels of FPGS activity that, in turn, facilitate enhanced intracellular formation of more cytotoxic MTX PGs.
A study involving 52 children with B-cell ALL did not confirm that MTX accumulation and polyglutamation have prognostic significance in patients receiving prolonged oral MTX therapy.139 This finding supports the notion that, under the conditions of continuous low dose drug exposure, the activity of MTX may not depend on cellular polyglutamation to sustain intracellular levels and antitumor effect. The role of PG formation in solid tumor therapy with MTX has been addressed in very few studies. In specimens from patients with soft tissue sarcoma, 12 of 15 tumors exhibited impaired polyglutamation, although the relationship to clinical response was uncertain.140,141
FIGURE 8-4 Methotrexate polyglutamate (MTXPG) content (p moles/109 bone marrow ALL cells) of subtypes of ALL following treatment in vivo with 1 g/m2 MTX over 24 hours. Hyperdiploid B-cell ALL (BHD), nonhyperdiploid B-cell (BNHD), ALL with E2A-PBX1 fusion, T-cell ALL, and ALL with TEL-AML1 fusion are subsets. One hundred-and-one patients’ samples were studied. Medians, quartiles, and ranges excluding outliers are shown, and P values from pair-wise comparisons (Wilcoxon rank sum test, adjusted for multiple tests) are given. (From Kager L, et al. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J Clin Invest 2005;115:110-117.)
The role of genetic variation in determining function of FPGS is suggested by the finding that two variants (R424C and S457F) slow the rate of PG formation for both folates and MTX and decrease cytotoxicity of MTX when expressed in Aux B1 cells.142
In summary, limited experimental and clinical evidence suggests, but does not conclusively prove, a relationship between clinical response and FPGS or GGH activity. The role of genetic variation in contributing to FPGS and GGH activities and PG formation remains to be defined.
Binding to Dihydrofolate Reductase
The physical characteristics of binding of NADPH (reduced form of nicotinamide adenine dinucleotide phosphate [NADP]) and MTX to DHFR of various species have been established by x-ray crystallographic studies, nuclear magnetic resonance spectroscopy, amino-acid sequencing of native and chemically modified enzyme, and site-directed mutagenesis, using enzymes from microbial, chicken, and mammalian sources143, 144, 145, 146, 147, 148; strong amino acid sequence homology is found at positions involved in substrate cofactor and inhibitor binding.149 In general, a long hydrophobic pocket binds MTX and is formed in part by the isoleucine-5, alanine-7, aspartate-27, phenylalanine-31 (Phe-31), and phenylalanine-34 (Phe-34), and other amino acid residues. Several particularly important interactions contribute to the binding potency of the 4-amino antifolates: (a) hydrogen bonding of the carbonyl oxygen of isoleucine-5 to the 4-amino group of the inhibitor; (b) a salt bridge between aspartate and the N-1 position of MTX, which is not involved in binding to the physiologic substrates; (c) hydrophobic interactions of the inhibitor with DHFR, particularly with Phe-31 and Phe-34; (d) hydrogen bonding of the 2-amino group to aspartate-27 and to a structurally consistent bound water molecule; and (e) hydrogen binding of the terminal glutamate to an invariant arginine-70 residue. Investigations have identified the importance of the interactions of MTX with Phe-31 and Phe-34 because mutations in these positions result in a 100-fold and 80,000-fold decrease in MTX affinity for the enzyme, respectively.150 Mutation of arginine-70 results in a decrease in MTX affinity by greater than 22,000-fold but does not alter the binding affinity of trimetrexate, which lacks the terminal glutamate moiety involved in this interaction.151 Mutations outside the enzyme active site also may result in marked reductions in folate and antifolate affinities.152 In addition, the physiologic substrate dihydrofolate is bound to the enzyme in an inverted, or “upside down,” configuration compared with the inhibitor MTX.147,153 The reader is referred to more detailed reviews of this subject for consideration of substrate and cofactor binding characteristics and studies of mutated DHFR.145, 146, 147, 148,154, 155, 156, 157
Optimal binding of MTX to DHFR depends on the concentration of NADPH. NADH (reduced form of nicotinamide adenine dinucleotide) may also act as a cosubstrate for DHFR but, unlike NADPH, it does not promote binding of MTX to the enzyme.158 Thus, the intracellular ratios of NADPH/NADP and NADPH/NADH may play an important role in the selective action of MTX to the extent that the cosubstrate ratios may differ in malignant and in normal tissues.123,158 In the presence of excess NADPH, the binding affinity of MTX for DHFR has been estimated to lie between 10 and 200 pM,159,160 although this affinity is significantly affected by pH, salt concentration, and the status of enzyme sulfhydryl groups. Under conditions of low pH and with a low ratio of inhibitor to enzyme, binding is essentially stoichiometric, that is, one molecule of MTX is bound to one molecule of DHFR.
Binding of MTX to DHFR isolated from parasitic, bacterial, and mammalian sources in the presence of NADPH generates a slowly formed ternary complex. The overall process has been termed slow, tight-binding inhibition and involves an initial rapid but weak enzyme-inhibitor interaction followed by a slow but extremely tight-binding isomerization to the final complex.145,157,161 The final isomerization step probably involves a conformational change of the enzyme with subsequent binding of the para-aminobenzoyl moiety to the enzyme.148 Other folate analogs, such as aminopterin, follow the same slow, tight-binding kinetic process, in contrast to the pteridines and pyrimethamine, which behave as classic single step inhibitors of the bacterial enzymes. Trimethoprim is considered to be a classic, albeit weak, inhibitor of mammalian DHFR. Of note, it does not undergo an isomerization process to the ternary complex form.157
In the therapeutic setting, and in intact cells, MTX acts as a tight-binding but reversible inhibitor. Under conditions of high concentrations of competitive substrate (dihydrofolate) and at neutral intracellular pH, a considerable excess of free drug is required to fully inhibit the enzyme. As drug concentration falls below 10−8 M in tissue culture and at lower concentrations in cell-free systems, enzyme activity resumes, and tritium-labeled MTX bound to intracellular enzyme can be displaced by exposure of cells to unlabeled drug or dihydrofolate,7,162,163 or reduced folates such as leucovorin and 5-methyltetrahydrofolate,123 which indicates a slow but definite “off rate” or dissociation of MTX from the enzyme.123,164 Thus, an excess of free, or unbound, drug is required to maintain total inhibition of DHFR.165
MTX PGs have similar potency in their tight-binding inhibition of mammalian DHFR101,157,166 and possess a slower rate of dissociation from the enzyme than the parent compound. In pulse-chase experiments using intact human breast cancer cells, MTX pentaglutamate was found to have a dissociation half-life of 120 minutes compared with 12 minutes for the parent compound. Cell-free experiments using purified preparations of mammalian enzyme indicate that MTX polyglutamation has a modest effect in enhancing binding and catalytic inhibition (twofold to sixfold) of DHFR.12,157,160,167 As with MTX, enzyme-bound MTX PGs may also be displaced by reduced folates123 and high concentrations of dihydrofolate,168,169 albeit at a slower rate than MTX.
These observations indicate that, in the absence of free drug, a small fraction of intracellular DHFR, either through new synthesis or through dissociation from the inhibitor, becomes available for catalytic activity and is adequate to allow for continued intracellular metabolism. The requirement for excess free drug to inhibit enzyme activity completely is important in understanding the clinical effects and toxicity of this agent and is fundamental to the relationship between pharmacokinetics and pharmacodynamics.
Resistance to MTX as a result of decreased DHFR binding affinity for MTX has been described in murine leukemic cells,152,170,171 Chinese hamster ovary172 and lung173 cells, and murine and hamster lung fibroblast cells.174,175 These mutant enzymes may have several 1,000-fold reduced binding affinity for MTX and, in general, are less efficient in catalyzing the reduction of dihydrofolate than is wild-type DHFR.
Drug-sensitive Chinese hamster lung cells express two different forms of DHFR encoded by distinct alleles.176,177 The two species differ in molecular weight and isoelectric point (21,000 versus 20,000 and 6.7 versus 6.5), a difference that results from a single amino acid substitution of asparagine for aspartic acid at position 95. Either allele may be predominantly expressed in various subclones of the parent cell line. This observation raises the possibility that distinct naturally occurring DHFR alleles may exist in a variety of tissues and, to the extent that they may confer differential sensitivity to MTX, this DHFR genetic polymorphism of the host may serve as a mechanism by which cells may become clinically MTX resistant.
DHFR with reduced affinity for MTX may represent a clinically important mechanism of MTX resistance, as this phenomenon was observed in the leukemic cells of 4 of 12 patients with resistant AML.178 Transfection of MTX-resistant, mutant DHFR has become a tool for creating drug-resistant hematopoietic progenitor cells and several laboratories have developed vectors to efficiently transduce human progenitor cells with the hope that such technology could be used to enable the use of higher, and hopefully more effective, doses of chemotherapeutic agents to treat human maligancies.179, 180, 181
A common finding in both laboratory and clinical studies of MTX-resistant cells is an increase in the expression of DHFR protein with no associated change in the enzyme’s affinity for MTX. Elevations in DHFR may persist for many generations of cell renewal in tumor cells from resistant patients. In resistant murine leukemic cells, the increased DHFR activity results from reduplication of the DHFR gene (Fig. 8-5), a process that occurs by exposing murine and human leukemia and carcinoma cells in culture to stepwise increases in the concentration of MTX.173,176,182, 183, 184 Gene duplication may take the form of a homogeneously staining region (HSR) on chromosomes or nonintegrated pieces of DNA known as double-minute chromosomes (DMs) (Fig. 8-5). Although HSRs appear to confer stable resistance to the cell, double-minute chromosomes are unequally distributed during cell division,175,183 and in the absence of the continued selective pressure of drug exposure, the cells revert to the original low-DHFR genotype. Evidence exists that gene amplification occurs initially in the form of doubleminute chromosomes because this is the predominant abnormality in low-level drug-resistant cells, whereas HSRs occur in highly resistant cells that contain multiple gene copies.175,183,184 Other investigations suggest the opposite sequence wherein chromosomal breaks result in HSRs, which are then processed to DMs or not, depending on how different cell types handle extra chromosomal sequences.185 Another mechanism of gene amplification has been identified in an MTX-resistant HeLA 10B3 cell line in which were found submicroscopic extrachromosomal elements (amplisomes) containing amplified DHFR genes. These amplisomes appeared early in the development of MTX resistance and were not found to be integrated into the chromosome, nor were they associated with double-minute chromosomes. Although these amplisomes were lost in the absence of the selective pressure of MTX, they disappeared at a much slower rate than would be predicted from simple dilution of nonreplicating elements.
Although MTX resistance through DHFR gene amplification becomes apparent only after the prolonged selective pressure of drug exposure, highly MTX-resistant cells may be generated by gene amplification within a single cell cycle.186 Early S-phase cells exposed transiently to agents that block DNA synthesis (e.g., hydroxyurea) may undergo reduplication of multiple genes synthesized during early S phase, including DHFR, after removal of the DNA synthetic inhibitor. This finding has broad implications for the rapid development of drug resistance in patients treated with MTX and other inhibitors of DNA synthesis. Exposure of cells to a variety of chemical and physical agents unrelated to MTX including hypoxia, alkylating agents,187 ultraviolet irradiation,187,188 phorbol esters,187
Only gold members can continue reading. Log In or Register to continue