Figure 8-1 Model organisms (the budding yeast—Saccharomyces cerevisiae; the fruit fly—Drosophila melanogaster; and the zebrafish—Danio rerio) and their key attributes as models for dissecting the molecular basis of cancer.
Forward Genetics, Reverse Genetics, and Transgenesis
Simple model organisms, with their rapid development and large numbers, lend themselves well to the identification of new oncogenes, tumor suppressors, and novel therapeutic targets important in cancer. This is generally accomplished using classical (and variations on classical) genetic screens. Forward genetic screens are based on Mendelian inheritance of genes and the observation of a phenotype in cells or organisms where a gene has been disrupted (Figure 8-3 ). The methods by which genes are disrupted and the assays used to screen for phenotypes are wide ranging, and some are species specific. Early screens looked for naturally occurring phenotypes of simple processes, such as inability of yeast to grow at a certain temperature, as a method of assaying genes involved in cell division. To increase the number of mutated organisms (mutants) with a phenotype of interest, methods of inducing mutations in genes, either chemically with retroviruses or with transposons (“jumping genes”), have been employed. Large numbers of organisms are screened to ensure that every gene within the genome has been mutated at least once—this is known as saturation of the genome. Forward genetic approaches allow unbiased isolation of phenotypically mutant organisms, after which the gene causing the mutant phenotype can be identified. The forward genetic screen remains the most powerful strength of model organisms as a cancer model, in particular for the discovery of novel tumor suppressors. Tumor suppressors are genes whose normal functions are to regulate uncontrolled or abnormal growth, or the growth of cells whose genetic integrity has been compromised. Because the cancer-causing effects of these genes can only be fully recognized when they are absent or nonfunctional, forward screening of large numbers of simple organisms is an attractive means to identify such genes in vivo. Historically, a potential drawback of such forward screens, particularly in the zebrafish, where there is somewhat greater genetic complexity, is that identification of the genetic mutation causing the phenotype can sometimes be time consuming and technically challenging: something of a “needle in a haystack” search. This is especially true if the mutation occurs in a regulatory or noncoding region of the genome or in telomeric regions of chromosomes where increased amounts of crossing over during meiosis further hamper conventional positional cloning techniques. However, the past decade has seen the emergence of massive parallel sequencing technologies that are now being used to facilitate rapid fine mapping of genetic mutants derived from phenotypic screens. 8,9 These technologies allow hundreds of genes to be sequenced simultaneously, covering the entire genome in such a way that regions of the genome linked to the mutant phenotype can be identified when the fish have been crossed to a strain of a different genetic background. The rapidly decreasing costs and increasing efficiency and availability of these technologies will likely fuel a new wave of studies in coming years, accelerated by advances in deep sequencing for positional cloning.
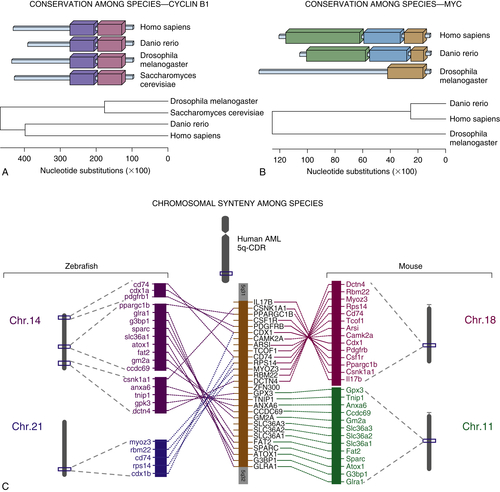
Figure 8-2 Conservation among species for (A) cyclin B1 and (B) the Myc oncogene. Each colored block represents a conserved protein domain. The phylogenetic trees below demonstrate common ancestries and the approximate number of nucleotide substitutions that have occurred leading to the divergence of the proteins for each of the given species. (C) The syntenic relationships among zebrafish chromosome 14 and 21, mouse chromosomes 18 and 11, and the region on the long arm of human chromosome 5 that is found to be critically deleted in myelodysplastic syndromes and acute myeloid leukemia with isolated loss of chromosome 5. AML, Acute myeloid leukemia; CDR, critically deleted region.
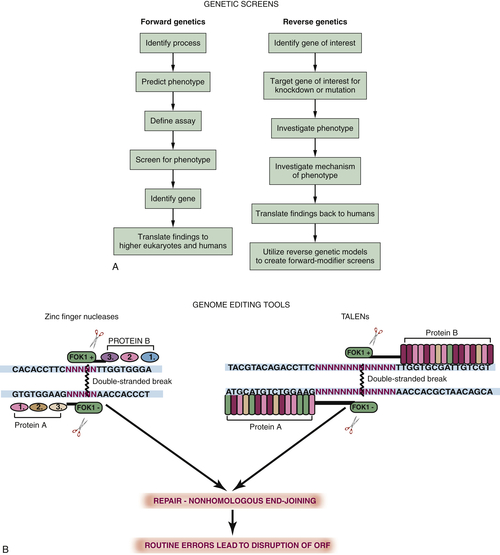
Figure 8-3 (A) Flow diagram illustrating the steps involved in forward and reverse genetic screens. (B) Schematic of the mechanism by which zinc finger (left) and tal effector endonucleases (right) function as genome editing tools. Zinc fingers are composed of two half-sites with three separate zinc finger domains in each. Each triplet is fused to the cleavage portion of the chimeric endonuclease FokI. Binding of the zinc fingers to the DNA target site results in FokI alignment for dimerization and cleavage of double-stranded DNA. Tal effector nucleases function in a similar manner. Each half-site is made of 14-16 tal effector repeats. Depending on two critical residues, these repeats will bind an A, C, T, or G, allowing a high level of specificity of DNA binding and subsequent ligation. Repair of the double-stranded break by nonhomologous end joining results in the introduction of insertions, deletions, or substitutions that result in mutant protein generation. ORF, Open reading frame; TALENs, transcription activator–like effector nucleases.
In addition to forward genetic modeling, it is also possible to disrupt specific known genes (such as known oncogenes or tumor suppressors) to investigate the phenotype produced. This process is termed reverse genetics. The most common reverse genetic studies are carried out using transient knockdown techniques. These include RNA interference (RNAi) in flies and antisense oligonucleotides (“morpholinos”) in frogs and fish. These techniques are particularly useful in determining epistatic relationships among genes giving rise to a particular phenotype, as several genes can be knocked down simultaneously. More recently, there have been a number of advances in the field of genome editing technologies that have made the generation of stable knockout lines at virtually any site within the genome accessible to researchers using simple organisms. 10,11 This is beneficial because it allows specific domains within a protein to be targeted for mutation and allows propagation of mutations through the germline for studies in later stage juveniles or adult organisms. This is achieved by the creation of synthetic restriction endonucleases that fuse the cleavage portion of FokI, a naturally occurring bacterial restriction endonuclease, to a site-specific DNA binding domain generated using synthetic zinc fingers (ZFNs) or transcription activator–like effector nuclease (TALEN) repeats. Two DNA sequences are targeted, one on either side of the region of the genome where a mutation is desired (see Figure 8-3, B). Because FokI requires dimerization to induce cleavage at its target site, this produces site specificity and minimizes off-target mutations. Novel technologies allowing the generation of large numbers of TALENs are likely to make the use of this form of genome editing widespread not only in simple organisms but also in mammalian models and cell culture systems.
Forward and reverse genetic models may be used in combination. Reverse genetics in simple organisms may add tools for the investigation of the genetic interactions of a known gene by permitting the development of modifier screens where the mutant is subjected to forward genetic screening to identify genes that enhance or suppress its phenotype. In this way modifier screens may provide novel therapeutic targets in human cancers.
Another genetic tool used in simple model organisms is transgenesis. To obtain a transgenic organism, specific DNA sequences are typically introduced into the genome of an organism and expressed under the control of a specific promoter sequence to guide the cellular, temporal, and spatial localization of transgene expression. For example, in zebrafish, the introduction of the mouse Myc gene under the control of the rag2 lymphocyte-specific promoter leads to expression of Myc in the thymus and the subsequent development of T-cell leukemia/lymphoma. 12,13 Adding the coding sequence of green fluorescent protein to the integrated construct has the additional advantage of permitting spatiotemporal visualization of the development of cancer in this system. A prime example is the recent use of dopamine β-hydroxylase promoter to drive MYCN and activated ALK expression in transgenic zebrafish to produce neuroblastoma, 14 demonstrating the power of this model for detailed studies to reveal cellular mechanisms underlying synergy between oncogenes that are also activated together in human malignancies. Transgenic approaches are easily performed in yeasts, which undergo efficient homologous recombination into their chromosomes, allowing site-specific integration of the transgene. Using this tool in yeasts facilitates reverse genetics by replacing the normal functioning gene with a mutated or nonfunctional form (which may be genetically engineered or even derived from a different species).
Drug Screens
Simple organisms can also be used for drug discovery in a variety of ways. Yeast can be manipulated to express a gene or protein of interest at a higher level than normal, or a heterologous human gene under control of a yeast-specific promoter. A differential effect of a drug on the normal-versus-mutated gene can also be assessed (e.g., attempting to find drug targets selective for certain oncogenes). 15 More recently, drug screens using whole organisms, including both flies and zebrafish, have been successful in uncovering potential novel therapeutic anticancer drugs and for revealing previously unsuspected anticancer activities of existing drugs. 16,17
Conditional Models
Only a small number of human cancers can be attributed to the presence of an underlying inherited cancer predisposition mutation; thus the majority of human cancers arises from a series of somatic mutations acquired over time. Because many human oncogenes and tumor suppressors play a critical role in embryologic development, germline mutations affecting key genes of this type in all tissues frequently lead to death during development. Although investigation of the embryologic phenotype in model organisms continues to provide crucial information on gene function, the study of genetic interactions in specific tissues and in specific cancer models provides additional information on how the mutated gene contributes to tumorigenesis in vivo.
Over the past 20 years, genetic tools have been developed to engineer targeted conditional expression or (in some cases) knockout of a specific gene. There are three main conditional systems that facilitate directing gene expression to a specific cell type at a specific time. The first is based on an enzyme from P1, which is a bacteriophage that infects the bacterium Escherichia coli. This virus produces an enzyme called Cre recombinase that cuts DNA whenever it sees two identical 34-base-pair sequences known as Lox-P sites. The enzyme removes the DNA between the two Lox-P sites, and the sites are ligated together. In model organisms, this system can be used by driving the expression of Cre with a promoter that is only expressed at a certain site (tissue specific) or with a promoter that is activated by exposure to a specific stimulus such as heat shock or a particular drug (e.g., estrogen). Lox-P sites can be introduced into a transgene such that on Cre activation, the transgene is expressed (or removed) in a specific tissue. Fluorescent proteins can also be incorporated into transgenes to allow visualization of where the transgene is expressed and where the Lox-P sites have been removed (Figure 8-4 ). In a similar fashion, Flp recombinase is an enzyme made by the 2-μm plasmid of Saccharomyces cerevisiae. This recombinase acts in a similar way to Cre recombinase, recognizing two 34-base-pair sequences termed Frt sites. This system has been extensively employed in flies, where instead of simply excising the intervening DNA sequence between two Frt sites, Flp recombinase results in the crossing over and exchange of genetic material between arms when Frt sites are located on opposite arms of the same chromosome. This allows, for example, the expression of mutant tissue in an otherwise wild-type background 18 (see Figure 8-4). Tissue-specific overexpression of a gene can also be achieved by using the yeast transcription factor Gal4 driven by a tissue-specific promoter and its upstream activating sequence (UAS) driving the gene of interest. UAS can also drive a fluorescent protein–colored marker to allow spatial localization of cells expressing the gene of interest (see Figure 8-4).
Yeast
Genetic Tools and Functional Genomics
Recovery of the genes responsible for a mutant phenotype observed in a forward genetic screen and harnessing the power of reverse genetics is made possible in yeast by the ease with which the organism can be transformed with DNA using plasmid vectors or polymerase chain reaction (PCR) products and by the subsequent ability of the yeast to undergo the process of homologous recombination. Homologous recombination is a conserved DNA repair process that allows recognition of homologous DNA sequences at sites of double-stranded DNA breakage. This recognition allows double-stranded breaks to be repaired by using the other intact copy (located on the sister chromosome) as a template. This physiological mechanism is hijacked when vector-derived DNA is designed to carry homology arms with sequences identical to the genome location where incorporation is desired. The yeast homologous recombination machinery then recognizes the homology arms and inserts the vector DNA sequence (chosen by the researcher) into the desired location. In this way, any gene can be replaced by another stretch of DNA. This could be a mutated or null allele or a normal allele to replace a mutated one. In addition, mutant or wild-type alleles can be coupled with genes encoding positive or negative selection factors to allow the identification of yeast strains that have undergone homologous recombination (such as an antibiotic or growth factor requirement). The application of these techniques is widespread, including the development of targeted gene “knockouts” such as those used in the yeast genome deletion project. An example of this is shown in Figure 8-5 . Molecular profiling of the effects of different growth conditions has been facilitated by the addition in the yeast genome deletion project of two unique 20-nucleotide tags that flank each knockout cassette. Using these tags as “barcodes” and starting with equal quantities of each deletion strain, gene chips have been developed to hybridize and quantify each tag (and thus knockout strain) in any given growth condition. 19
Because the yeast genome is relatively small and contains little noncoding DNA, it has been possible to derive plasmid libraries for all genes found in yeast that are of a manageable size. This is done by cutting the whole genome with restriction enzymes that recognize a particular DNA sequence and then inserting each of these smaller cut pieces of DNA into a plasmid vector. These DNA libraries are useful to identify the gene causing a phenotype or to address the phenotype resulting from the overexpression of certain genes (e.g., which can also be used in a drug screen).
One complication of examining genotype-phenotype relationships is that for some genes loss of function and/or gain of function do not show a phenotype. The likelihood is that these genes are either redundant or act in pathways whose physiological functions are buffered by other cellular pathways acting in parallel. 20 To decipher the interaction between two deleted genes (synthetic fitness or, more severely, synthetic lethality when the interaction causes lethality) or between a deleted gene and an overexpressed gene (known as synthetic dosage fitness or lethality), novel synthetic genetic array (SGA) screens have been employed. 21,22 Yeasts with the mutation of interest in the haploid state are mated to a pool of haploid yeast knockout strains of the opposite mating type. Selection markers are introduced to allow identification of double mutant strains, and their phenotype is assessed. Synthetic lethality screens have been further developed to incorporate the microarray technology using barcodes as described earlier—the synthetic lethality analyzed by microarray (SLAM). 20 SLAM has allowed analysis of double-mutant heterozygous diploid strains of yeast, as well as other genetic interactions such as chemical genetic interactions. 23
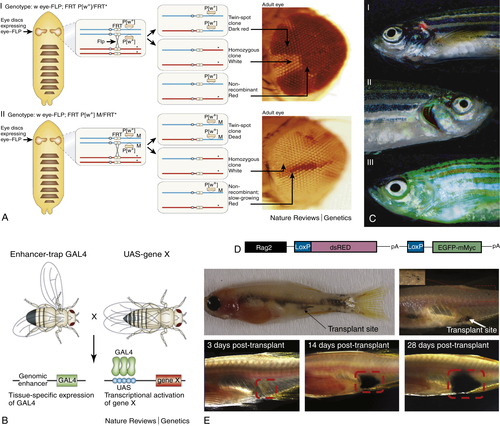
Figure 8-4 (A) Flt/FRP system/twin-spot clones. By placing the FLP recombinase gene under the control of the eyeless enhancer (which drives expression specifically in the eye-antennal imaginal disc), Flp/FRT-mediated recombination can be targeted to this disc to generate homozygous mutant clones in the eye in flies that are otherwise heterozygous. (I) The non-mutant chromosome (the asterisk indicates a mutation) is marked by a mini-white transgene, but there is no selection against the twin-spot clones or nonrecombinant cells, and both the mutant clones (white) and the twin-spot clones (darker red, because they carry two copies of white+) are relatively small. (II) The effects of incorporating a Minute mutation (M) onto the nonmutant FRT chromosome. The mutant clones now occupy almost all of the eye, because they outcompete the slow-growing nonrecombinant cells (which are M/+), whereas the twin-spot clones die. (B) Gal4/UAS system. The yeast transcriptional activator Gal4 can be used to regulate gene expression in Drosophila by inserting the upstream activating sequence (UAS) to which it binds next to a gene of interest (gene X). The GAL4 gene has been inserted at random positions in the Drosophila genome to generate “enhancer-trap” lines that express GAL4 under the control of nearby genomic enhancers, and there is now a large collection of lines that express GAL4 in a huge variety of cell-type and tissue-specific patterns. Therefore, the expression of gene X can be driven in any of these patterns by crossing the appropriate GAL4 enhancer-trap line to flies that carry the UAS–gene X transgene. This system has been adapted to carry out genetic screens for genes that give phenotypes when misexpressed in a particular tissue (modular misexpression screens). (C,D) Cre-Lox system. Zebrafish carrying the construct shown in (D): (I) No cre activation—fish show red thymus, showing that the dsRED cassette has not been excised. (II) Following cre injection, the dsred stop cassette is removed, allowing expression of the cMyc-EGFP fusion oncogene. This fish has an enlarged green thymus indicating the development of T-cell acute lymphoblastic lymphoma. (III) In this older fish, the whole fish fluoresces green, indicating disseminated T-cell acute lymphoblastic leukemia. (E) Tumor transplantation experiment using the transparent “casper” fish. These images show the development of melanoma (derived from p53 null/BRAFV600E model) over time in the same animal. The tumor can be monitored externally without sacrificing the fish. (A and B, Reprinted with permission from Macmillan Publishers Ltd: Nat Rev Genet. St. Johnston. C, Courtesy Hui Feng (unpublished data). E, Reprinted from White et al., Cell Stem Cell. Copyright 2008, with permission from Elsevier.)
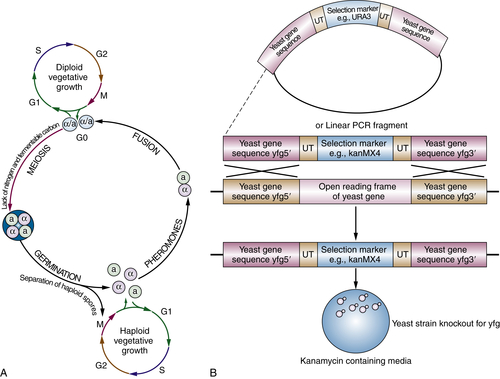
Figure 8-5 (A) A schematic representation of the budding yeast life cycle. Under adequate nutritional conditions diploid yeast cells undergo vegetative growth by mitosis (top left). The cells cycle through G1, S (synthesis), G2, and M (mitosis) phases. When starved of nitrogen and fermentable carbon, sporulation occurs with formation of gametes by meiosis. The gametes are contained within a casing called an ascus. As the gametes germinate, the haploid spores separate. The a and α gametes secrete pheromones leading to fusion of an a to α gamete when they meet. If separated, however, haploid vegetative growth can also continue by mitosis (bottom right). (B) Schematic representation of replacement of yeast gene yfg (your favorite gene) with a deletion cassette containing the selection marker KanMX4 conferring resistance to the antibiotic G418 (kanamycin). 40-50 bp of yfg gene sequence at the 5′ and 3′ ends are cloned with KanMX4 selection marker between, flanked on each side by 20-bp unique tags (UT). The yfg sequences are recognized by the homologous recombination machinery in the yeast, and a proportion of yeasts will swap the genetic material encoding the gene for that encoding the selection marker. Yeasts that survive in media containing kanamycin have incorporated the deletion cassette and no longer contain a functional yfg. These strains can be isolated from mixed cultures by PCR using the flanking UT. (Adapted by permission from Macmillan Publishers Ltd: Nat Rev Genet. Kumar A and Snyder M. Emerging technologies in yeast genomics. Copyright 2001.)
Yeast screens have been devised to determine not just genetic but also protein-protein interactions. These can be in the form of two-hybrid screens, or more recently in the form of tandem affinity purification tagged (TAP-tag) analysis. The principle of the two-hybrid screen analysis is that transcription factors require two domains—a DNA binding domain (BD) and an activation domain (AD) in close proximity to one another—in order to bind to and cause transcription of their target gene. Part of the Gal4 transcription factor (either the BD or the AD; for this example the BD) is fused to the protein of interest, for example, the MYC oncoprotein. A library of other proteins is then fused to the Gal4 AD. If any of the library proteins interacts with MYC, then the Gal4 transcription factor will bind and cause transcription of target genes. The target gene can be engineered to be a selection factor that will only allow yeast to grow in the presence of its expression in selective culture conditions, and its identity can be confirmed by subsequent sequencing. 24 In TAP-tag analysis, a tandem affinity purification protein tag is introduced into the open reading frame (ORF) of each yeast gene to create the expression of a fusion protein. This protein tag is specially designed to allow efficient affinity purification of the tagged protein of interest along with other proteins bound or associated with it. The resultant interacting proteins can then be resolved by a variety of methods, the most accurate of which is mass spectrometry. 25,26
The Cell Cycle
The fundamental processes of cell growth and division are governed by the tightly regulated processes that maintain the cell cycle. The cell cycle is an ordered set of events by which cells grow and divide to produce two identical daughter cells. It is divided into four phases, as shown in Figure 8-5. Two features of cancer cells in mammalian systems are controlled (at least in part) by cell cycle maintenance: altered growth and genomic instability. Based on a number of features, yeasts provide the ideal system for study of the cell cycle. 27 The budding yeast Saccharomyces cerevisiae has been the most highly investigated. S. cerevisiae is able to survive in both haploid and diploid states. In the presence of sufficient cell nutrients, diploid cells undergo division by mitosis and growth by budding. The size (or presence) of a bud can be visualized by light microscopy and permits determination of cell cycle status in individual cells. Each diploid cell contains a copy of each of the two mating types—Mata and Matα—determined by the MAT locus. When diploid cells are starved of nitrogen and fermentable carbon, they undergo sporulation and commence formation of a and α gametes. Here the cells undergo division by meiosis, followed by differentiation of the subsequent haploid progeny. The haploid progeny immediately fuse with a cell of the opposite mating type to produce a diploid cell—a process determined by a mating pheromone that is specific to each mating type (a or α). Therefore the cell cycle and mating machinery are integrally linked. 28,29 In addition, yeast cells that have arrested at different parts of the cell cycle (usually under specific conditions such as high temperature) have disrupted genes critical to progression through the cell cycle and cell growth, which may be potential oncogenes and/or tumor suppressors. The study of mutant yeast strains with abnormalities in the cell cycle has led to the identification of genes orthologous to human oncogenes and tumor suppressors.
CDC2 and CDC28
In 2001, the power of yeast as a genetic tool for studying the cell cycle and its implications in cancer were recognized with the award of the Nobel Prize in medicine to Paul Nurse and Leland Hartwell. They not only elucidated many of the critical genes involved in regulating the cell cycle but also demonstrated the genetic and biochemical conservation that makes study in this and other simple organisms so powerful. Two of the critical genes identified by these investigators continue today to provide insight into how cancer cells evade normal growth and cell cycle regulatory mechanisms. The CDC28 gene was discovered in the early 1970s by Leland Hartwell in an early genetic screen to identify S. cerevisiae strains that conditionally blocked cells at different stages of the cell division cycle (cdc). The presence of Cdc28 was found to be essential for cells to initiate both the nuclear and cytoplasmic events required for cell division. Before the Cdc28-dependent step, yeast cells are able to undergo either sexual replication or entry into the cell cycle, and thus the Cdc28 step in G1 came to be known as the start of the cell cycle in the budding yeast. The CDC28 gene was cloned in 1980, and by 1985 Cdc28 was shown to have protein kinase activity. 30
Paul Nurse uncovered a critical regulator of the cell cycle in S. pombe. Using the observation that in budding yeast, cell size is coordinated with progression through the cell cycle, a number of “wee” (meaning small) mutants were identified. “Wee” mutants progressed through the cell cycle more rapidly than normal before sufficient time for normal growth had passed, resulting in the generation of the small phenotype. The second “wee” mutant isolated, wee2, was shown to map the same locus as cdc2 and later, in 1982, was shown to be functionally homologous to the CDC28 gene in budding yeast. The cloning of the human CDC2 gene in 1987 confirmed the utility of this system for investigating the role of specific genes in humans. 27 In more recent years, cdc2 and CDC28 have been renamed CDK1 (cyclin dependent kinase 1) as part of the CDK family of kinases, which (along with other roles in cell cycle, transcription, and differentiation) associate with cyclins, allowing intricate control of the cell cycle. CDK1 associates in particular with cyclin B and is involved in the control of mitosis (reviewed in Refs. 31 and 32). Since then, abnormal expression of CDK1 has been found in a variety of human cancers and is now known to be required for efficient phosphorylation of the Bloom syndrome DNA helicase (BLM). 33 Homozygous mutation of the BLM leads to Bloom syndrome—a condition in humans that predisposes to multiple forms of cancer as a result of genomic instability.
Ploidy, Genome Instability, and Cancer
The study of Leland Hartwell’s temperature-sensitive cell division mutants led to the analysis of genes essential for cell cycle progression and their roles in determining the fidelity of genetic replication. Several cell cycle mutants demonstrated markedly increased rates of chromosome loss, recombination, or mutation. From this evidence, the DNA damage checkpoints and DNA repair pathways active during the cell cycle were first identified. One such mutant carried a mutation in POL3, a gene now known to encode the catalytic subunit of DNA polymerase subunit δ. This polymerase forms part of a proofreading enzyme whose role is to limit errors occurring during DNA replication by removing any incorrectly incorporated nucleotides via intrinsic exonuclease activity. Homozygous mutations in POL3 are embryonic lethal (in yeasts and mammals), but heterozygous mutations within the catalytic domain result in a mutator phenotype. The mutator phenotype—where a mutation in one gene predisposes to the development of additional mutations—is known to contribute to the development of cancer in mammals, and thus heterozygous mutations in Pol3 result in increased rates of cancer in mice. However, in bacteria it is known that the presence of a mutator phenotype results in reduced fitness of the bacterium. This occurs because with each division, an increase in the number of deleterious mutations occurs, ultimately reducing its ability to grow and divide. This continues until such time as an “antimutator” mutation occurs that decreases the number of de novo mutations, allowing this subpopulation to have the growth advantage. It is possible that such a process also occurs in humans, contributing to the clonal evolution of tumors over time. Herr et al. recently tested whether this phenomenon was active in eukaryotic cells using budding yeast, with the hypothesis that a critical threshold of mutations could be tolerated before a yeast became incapable of further cell division. 34 The authors identified several mutations within conserved residues of Pol δ that exacerbated the mutator phenotype observed in a yeast strain also carrying a mutation within the mismatch repair gene MSH6 (msh6Δ). In addition to confirming their hypothesis that a threshold number of mutations can be tolerated within a eukaryotic cell, the authors identified occasional yeast colonies that grew in the presence of apparently otherwise synthetic lethal gene mutation combinations. Assessing these “error-induced extinction” (eex) mutants, they determined that indeed these yeasts had developed antimutator phenotypes, with a third of cases having additional antimutator mutations within the POL3 gene itself as the mechanism of their escape. This elegant study highlights evidence that supports some recent clinical observations in cancer, such as the remarkable success of inhibition of the poly(ADP) ribose enzymes (PARP). PARP1 and PARP2 enzymes are an integral component of the repair of single-stranded breaks as cancer therapies. In the presence of mutations in genes involved in homologous recombination, such as BRCA1 and BRCA2, found in breast cancers among others, PARP inhibition results in selective tumor killing. 35 The likely mechanism of this is the synthetic lethality of tumor cells that arises from the breaching of the critical threshold of mutations sustainable by an individual cell. However, it does raise the concern that the clonal evolution of cells resistant to this mechanism of killing is likely to be problematic when using this therapeutic strategy in some cases.
Flies
Why Use a Fly?
Drosophila melanogaster has been used as a model organism in developmental biology and genetics for a century, and the resultant generation of a vast array of tools make it one of the most comprehensively genetically tractable systems for study. Several features of the fruit fly have led to its popularity as a genetic model, most critically the small number of genes contained within the four fly chromosomes, relatively little redundant DNA, and large numbers of human homologs that have been identified following the complete sequencing of both the fly and the human genomes. In addition, fruit flies develop large polytene chromosomes in the salivary gland. These chromosomes are produced in the last larval stage (the third larval instar) when large amounts of glue proteins are required for pupation. The large amount of protein production is achieved by genome amplification by a process called endoreduplication—DNA replication without division. When stained by standard G-banding, the resolution of endoreduplicated chromosomes is an order of magnitude greater than that seen of human chromosomes, as there are multiple copies of each gene. This in turn facilitates the identification of genes that have been deleted. Although wild-type flies do develop tumors, the similarities of these spontaneous growths to mammalian cancers are limited. However, forward genetic analysis has uncovered cancer-like proliferations in the developing fly larva that provide an excellent platform for the investigation of tumorigenesis in this multicellular organism. Early in embryogenesis, cell fate is assigned, leading to the formation of certain adult structures, which develop in the larva via saclike invaginations of specialized epithelium (known as imaginal discs). There are 15 imaginal discs—seven bilaterally symmetrical pairs and one germ cell imaginal disc. Imaginal discs consist of a single layer of cells that can be easily visualized in the developing larva. In addition, both brain and blood cell neoplasia can be seen in mutant fruit flies. 36 Not all aspects of human cancer can be modeled in a fly, however. In particular, flies lack a closed vascular system, and thus angiogenic properties of tumors cannot be investigated. Despite this, genes known to regulate the angiogenic properties of human tumors, such as vascular endothelial growth factor (VEGF), have fly homologs that have been implicated in tumor development in flies.
Another feature of the fruit fly that makes it an attractive model to study is the availability of large banks of mutant flies. Mutagenesis in flies has been performed using x-rays, chemical agents to induce point mutations, and P-element–mediated insertional mutagenesis. P-elements are transposons or sequences of DNA that can move around or “jump” within the genome. When transposons insert into the genome at the beginning of a coding sequence, that gene’s transcription is disrupted, generally creating a null allele. Identification of the disrupted gene is much easier than with a chemically induced point mutation, because the sequence of the P-element is known and can be “tagged,” and PCR primers can be used to facilitate gene identification. To determine which flies carry the P-element, its sequence can also be modified to carry a marker, such as rosy eyes. “Jumping” of the P-element requires the function of another gene, transposase, and thus the disrupted gene will be fixed unless the transposase is present. The transposase gene can be bred into flies carrying a P-element to induce the latter to move and is usually carried on what is known as a “balancer” chromosome with another mutation that is easily identifiable, such as curly wings.
Genetic Tools
The first tumor suppressors identified by forward genetics in flies exhibited only one or two features of cancer. The phenotypes observed were of hyperplasia or neoplasia in a single tissue affecting 100% of flies with the mutations and, unlike in human cancers, there did not appear to be a requirement for the development of additional mutations to cause tumorigenesis. Because these initial tumor suppressor genes were not homologous to those found in humans, the fly became less popular as a model organism to study cancer. However, this soon changed, as highly conserved signaling pathways were subsequently identified in flies and humans and more sophisticated genetic techniques to study gene interactions became available. 37 These included genetic screens to identify second-site modifiers of known tumor suppressors; the discovery of a Drosophila homolog of C-terminal src, dcsk, isolated by Stewart et al. in a screen for dominant modifiers of the lats tumor suppressor 38 ; and the dissection a number of second-site modifiers of the transcription factor E2F. 39,40
More recently, the use of the conditional Frt/Flp and Gal4/UAS systems has been invaluable in cancer research in the fly. Focusing on the fly’s greatest strength—that it offers the ability to investigate cell-cell interaction at a single-cell level—the introduction of mosaic clone analysis for the first time underpinned the role of the microenvironment and non–cell-autonomous cues in the life of an individual cell. 41 The utility of this tool has been used to dissect the cell-autonomous and non–cell-autonomous roles of the Drosophila myc gene. 42–44 Similarly, the interactions between oncogenes and tumor suppressors have been evaluated in mosaic clones, demonstrating cooperation between many known oncogenes and fly tumor suppressors. 45–48
The normal processes by which cells migrate during different developmental processes have been studied extensively in the fruit fly. Different processes use different modes of migration, requiring alterations in cell polarity, cell shape, and the adhesion of cells both to other cells and to the extracellular matrix. Disruption of genes and signaling pathways employed in normal migration processes has been shown to be involved in the ability of cancers to invade local tissues and metastasize to distant regions. This area of research is in its infancy, but the extensively delineated normal processes will undoubtedly assist in the investigation of mechanisms by which cancer cells evade their local environment in this model. 18
Malignant Neoplastic Tumor Suppressors in Drosophila: scribble and Others
Many fly mutant genes were classified as tumor suppressors in early embryonic screens for larval tissue overproliferation. However, subsequent characterization of a number of those genes demonstrated mechanisms of tissue expansion that did not represent features normally ascribed to cancer cells, and thus these genes are no longer considered true tumor suppressors. Despite this, the recent identification of the tumor suppressor scribble was through a screen designed to identify maternal effect mutations that disrupted aspects of normal epithelial morphology. 45 It was noted that scribble mutants, in addition to disrupting epithelial morphology in the embryo, also led to epithelial defects in the monolayer epithelium of the female germ cells (follicle cells), in which clones of mutant cells were expressed among wild-type cells. 45 A further screen for additional mutant clones affecting the follicle cell epithelium led to the identification of another mutant with a phenotype almost indistinguishable from that of the scribble mutants. Mapping of this mutation revealed it to be an allele of a previously identified tumor suppressor called lethal giant larvae (lgl). It was also noted that mutant clones of another known tumor suppressor, discs large (dlg), led again to a very similar phenotype in follicle cells. In normal tissues, the role of the Scribble protein is in maintenance of cell polarity and cell-cell adhesion, by controlling the localization of other proteins within epithelial cells in order to maintain correct spatial orientation. Bilder et al. postulated that given such similarities in phenotype scribble may be a tumor suppressor like lgl and dlg and also that lgl, dlg, and scribble might interact to form the necessary machinery to maintain cell architecture and cell proliferation in fly epithelium. 45 The identity of scribble as a tumor suppressor was confirmed by investigation of the epithelium of the third-instar larvae imaginal discs, which demonstrated cellular overproliferation with loss of apicobasal polarity and disordered architecture. In addition, overgrowth of brain tissue was observed in scribble mutants—another feature common to the lgl and dlg mutants. Epistatic relationships among the three genes have also been demonstrated by the ability to enhance the phenotype of scribble mutants by an additional heterozygous mutation of either dlg or lgl. Following the discovery of scribble and its properties as a tumor suppressor in flies, a further screen set out to use the fly as a method of studying the metastatic properties of tumors. Until this point, individual tumor suppressors and oncogenes that had been studied in flies apparently lacked the capability to proliferate without the microenvironment in which the malignant cells reside. This is perhaps not surprising, given the premise that a single genetic lesion is rarely sufficient to promote tumorigenesis, but that it creates a mutator phenotype predisposing to the additional mutations required for cancer to develop; and that in an organism such as the fly, the likelihood of that secondary mutation occurring is low within its short natural lifespan. The screen that was developed investigated the interaction of activated ras (rasv12) and known and unknown additional mutations. The system allowed for the development of clones of malignant cells that expressed ras plus additional mutations in the normal microenvironment within the eye disc, using the Frt/Flp system, the Gal4/UAS, and the Gal80 suppressor to localize expression. The results demonstrated that the combination of a ras v12 mutation and scribble mutation led to circulating tumor cells within the fly hemolymph open circulation and the development of widespread metastatic tumor formation. In these metastatic tumors, basement membrane integrity was breached (as in mammalian metastatic tumors), and overexpression of the junctional adhesion protein E-cadherin suppressed the metastatic behavior of the tumors. This is also in keeping with mechanisms of metastasis in human epithelial tumors, where E-cadherin is frequently downregulated. 50 These studies unequivocally demonstrate the utility of the fly as a cancer model with unique properties for uncovering novel genetic interactions and potential therapeutic targets. Recent studies on the human SCRIBBLE gene have shown that it is downregulated in cervical cancer 51,52 and the majority of invasive breast cancers 46 and interacts with the adenomatous polyposis coli gene, leading to altered expression in many cases of colon cancer. 53
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


