Abstract
Androgens are critical to the normal developmental processes in vertebrates, affecting the development of the normal male phenotype during embryogenesis and regulating a range of processes in adults. In normal development, the actions of two related steroids, testosterone and 5-alpha-dihydrotestosterone (DHT), are required for the development of the normal male phenotype. These steroid hormones bind to and modulate the activity of the androgen receptor (AR), a member of the steroid hormone receptor family that is encoded on the X-chromosome. The cloning of the human AR and the analysis of the AR genes of patients with various forms of androgen insensitivity have permitted numerous investigations that have yielded a wealth of information about the types of alterations of the AR gene that result in clinically evident alterations of AR function.
Keywords
androgen receptor (AR), 5-alpha-dihydrotestosterone (DHT), virilization, androgen insensitivity, DNA-binding domain (DBD), ligand-binding domain (LBD), Reifenstein’s syndrome, spinal and bulbar muscular atrophy
Androgens are critical to the normal developmental processes in vertebrates, affecting the development of the normal male phenotype during embryogenesis and regulating a range of processes in adults. In normal development, the actions of two related steroids, testosterone and 5-alpha-dihydrotestosterone (DHT), are required for the development of the normal male phenotype. These steroid hormones bind to and modulate the activity of the androgen receptor (AR), a member of the steroid hormone receptor family that is encoded on the X-chromosome. The cloning of the human AR has permitted a wealth of information about the types of alterations of the AR gene that result in alterations of AR function.
Male phenotypic development is controlled by androgens
The pioneering studies of Jost established the endocrine basis of male phenotypic development, and subsequent experiments have confirmed and refined this model ( Fig. 20.1 ). In mammals, the determination of whether the indifferent gonad will differentiate as a testis or an ovary is determined by the complement of sex chromosomes that are present. This fundamental developmental step determines the pattern of sex-specific secretion of gonadal hormones. In male embryos, testosterone is secreted by the Leydig cells of the testes beginning at approximately nine weeks of gestation. This steroid hormone acts to virilize the fetal Wolffian duct structures. Although the synthesis of testosterone is required for male sexual development, it is not sufficient for full male phenotypic development. The synthesis of DHT from testosterone by the 5-alpha reductase 2 enzyme is required for virilization of the male external genitalia and development of the prostate, conclusions supported by both human studies of patients deficient in the 5-alpha reductase 2 enzyme and the experimental use of inhibitors of the 5-alpha reductase 2 enzyme.
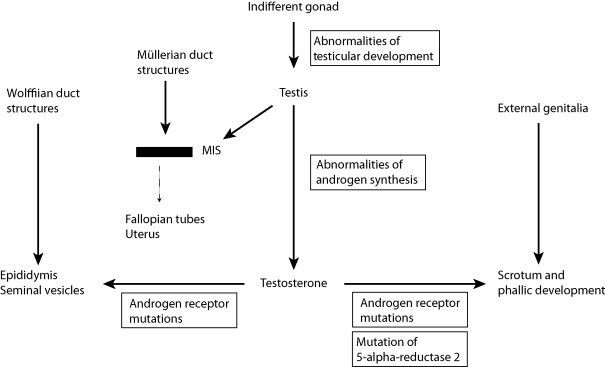
While this chapter is focused on the molecular defects of the AR, it is important to note that the Sertoli cells of the fetal testes also secrete a second hormone essential for normal male phenotypic development: Müllerian inhibiting substance. This polypeptide hormone, a member of the TGF-beta hormone family, acts to effect an involution of the embryonic Müllerian ducts, structures that persist and develop in the female embryo, giving rise to the uterus and fallopian tubes. Abnormalities of MIS synthesis or action underlie forms of male pseudohermaphroditism in which male phenotypic development has occurred (virilization of the internal and external genitalia has occurred normally), but in whom the uterus and fallopian tubes are present.
The androgen receptor
The AR was one of the first receptor proteins to be characterized following the synthesis of high specific activity androgen ligands. The cloning of cDNAs encoding the receptor protein permitted its identification as a member of a family of structurally related transcriptionally regulated transcription factors, the nuclear receptor (NR) family. Each family member contains specific structural motifs: an amino terminus of varying length, a DNA-binding domain (DBD) containing two “zinc fingers,” and a carboxyl-terminal ligand-binding domain (LBD) ( Fig. 20.2 ).
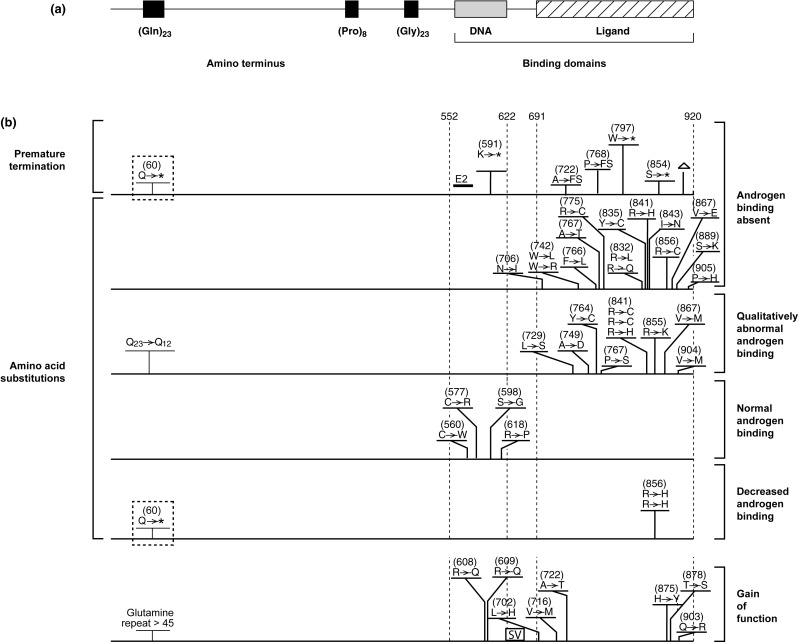
The central DBD is the most highly conserved element within each of the NR proteins. The sequence similarities of this distinctive motif were sufficiently high among the different members that nucleotide sequence homology between them permitted the cloning of several members of the NR family, including the AR. This region of each of the NR proteins encodes two short segments that contain highly conserved, specifically spaced cysteine residues that each coordinate a zinc atom (zinc fingers). In a number of naturally occurring mutations and in site-directed mutagenesis studies, these highly structured elements have been demonstrated to define the components of each receptor protein that are able to directly recognize and bind to specific DNA sequences within target DNA sequences within and surrounding target genes.
The region of the NR proteins responsible for the high-affinity binding of ligands is the LBD. This region is also highly conserved, albeit not to the degree observed between the DBD regions of the NRs. The overall structural homology of this segment is present, even among members of the family not known to bind a ligand. The LBD of the AR has been demonstrated to extend from approximately amino acid residue 690 to its carboxyl terminus, a size consistent with this segment in other members of the NR family. Structural studies have established the crystal structures of the AR and shown that its structure is similar to the organization of this region in other members of this family. The primary structure of the AR constitutes 12 alpha helices that are arranged in a three-layered sandwich around a central binding pocket. The position of the terminal helix relative to the remainder of the LBD is modulated by the binding of agonist ligands. The position of this region determines whether critical surfaces needed for the recruitment of receptor coactivators are exposed.
In contrast to the DBD and LBD of the AR, the functional role of the amino terminus of the receptor protein is less easily defined. This region is large, encompassing more than half of the primary amino acid sequence of the AR. In deletion experiments using androgen responsive genes to measure the function of the AR, it was observed that deletions of large segments of the AR amino terminus were required to substantially perturb AR function; no individual amino acids or small segments were shown to be critical for alter AR function. This may well indicate that the functional elements of the AR amino terminus are spread throughout this region in a redundant fashion. Alternatively, the requirement of large portions of the amino terminus for AR function may reflect its participation in facilitating the binding of DNA, ligand, and coactivators needed for full function. It is interesting to note that several lines of investigation have concluded that interactions between regions of the amino and carboxyl termini of the AR can influence one another and may exert discernible effects on AR function.
The amino acid sequence of the human AR contains three segments composed of repeated amino acid residues: one of glutamine, one of glycine, and one of proline. Similar repeat elements have been identified in other transcription factors, including other members of the steroid receptor family. The repeated proline motif does not appear to be constant, and variations in its length have not been reported. Similarly, polymorphisms of the glycine repeat appear to be relatively infrequent and of little biological consequence. These circumstances stand in contrast with the frequent polymorphisms of the glutamine repeat in the human population. Of note, differences in the length of this segment have been associated with subtle differences in AR function. Shortening of this same region has been associated with biological attributes such as infertility and a predisposition to prostate and breast cancer. Importantly, expansion of this region has been shown to cause spinal and bulbar muscular atrophy, the first of the “triplet expansion” diseases to be identified.
Measurements of AR and its function
Assays to detect the AR were first developed using high specific activity radiolabeled ligands to measure the high affinity binding of these ligands in target tissues and cell lines. These assays were used by several groups to characterize the physical properties of the binding protein is tissue extracts and in samples from cell lines established from normal subjects and from patients with known or suspected abnormalities of AR function. Monolayer binding assays permitted many such subjects to be identified as having abnormalities of the AR on the basis of alterations in the absolute amounts of AR measured or qualitative abnormalities of ligand binding. Although these assays were often informative, a significant number of patients displayed no differences of ligand binding, many times in circumstances where androgen insensitivity was highly likely (e.g., on the basis of family history).
Following the cloning of AR, the predicted amino acid sequence was used to generate antibodies to different regions of the AR protein. The most useful of these were those directed at the first 20 amino acids of the receptor proteins. These epitopes are highly conserved and are present in a number of mammalian species. While useful at the level of the research laboratory to categorize AR expression in patient samples, such assays have not been widely used clinically.
As noted previously, the AR is highly related to other members of the steroid receptor family, particularly the progesterone and glucocorticoid receptors. The definition of model genes responsive to the progesterone and glucocorticoid receptors in cell transfection assays was adapted to the study of the function of the AR. This provided methods to define the functional domains of the AR and to assess the impact of mutations of the AR on its function, independent of the ability to bind ligand. The delivery of such reporter genes directly into patient fibroblasts permitted the assay of the activity of the natively expressed receptor protein. In a subset of such samples, the activities were suggestive of defects in non-AR proteins important for AR function.
The modulation of gene expression by the AR
The human AR exerts its principal effects in the nucleus and the sequences within the AR responsible for nuclear localization sequences within and adjacent to the second zinc finger have been defined. In the absence of ligand, the AR exists in association with a complex of chaperone proteins. Following binding of agonist by the receptor, it undergoes extensive conformational changes, particularly the position of the terminal helix of the LBD. This change permits the binding of target DNA sequences and the recruitment of ancillary proteins that facilitate and stabilize interactions with complexes of proteins, including receptor coactivators, that interact with the basal transcription machinery. These alterations result in the binding of the activated AR to specific sites within the genome, alterations of chromatic structure, and changes in activity of androgen responsive genes. Distinct conformations of the AR LBD result when complexed to AR antagonists. These distinctive conformations recruit different classes of proteins (corepressors) that recruit distinct complexes that result in decreases in androgen responsive genes.
Androgen insensitivity: a spectrum of abnormalities caused by defects of the AR
Genetic defects of the AR are relatively common, a fact that can be explained by several features of the AR and of male phenotypic development. First, the AR gene is located on the human X chromosome, a chromosomal location that ensures that in genetic males, only a single copy of the AR gene is present and that a significant alteration of AR function will be translated into a discernible alteration of male development. Second, the detection of abnormalities of sexual development during the routine careful examination of neonates is often a trigger for extensive evaluations. Finally, while defects of AR function may have substantial effects on the development of the male phenotype, they appear to have limited impact on viability.
In early descriptions, it was not clear how the different phenotypes associated with abnormalities of male sexual development caused by defects of the AR related to one another. It was not clear whether patients with apparently different phenotypes such as complete androgen insensitivity (complete testicular feminization) and the Reifenstein syndrome were due to specific classes of mutations within the AR. It is now evident that androgen insensitivity is a spectrum, and the phenotype that is observed in an individual patient represents the degree to which the function of the AR proteins has been disrupted or the level of the AR expressed has been altered. In many instances these attributes (level of expression and function) are two intertwined aspects of the underlying genetic change that dictates the extent that the effects of androgen in male phenotypic development have been disturbed.
At one extreme of the phenotypic spectrum are individuals in whom the AR possesses no remaining function. Such alterations may include partial or complete gene deletions, premature termination, or an amino acid substitution in a critical domain of the receptor protein. As a result of this absence of AR function, none of the normal effects of testosterone and 5-alpha-DHT occur: phallic growth and scrotal formation do not occur despite the secretion of normal amounts of androgen. Instead of virilization, such individuals with complete androgen insensitivity display a feminized phenotype. The circulating levels of testosterone – although unable to effect virilization – are aromatized to estrogen, which stimulates normal breast development. The normal testes present in these individuals secrete MIS, which mediates the involution of the structures derived from the Müllerian ducts. As a result, these individuals do not have fallopian tubes or a uterus, the vagina is blind-ending, and the testes are either located within the abdomen or within the inguinal canals. A large number of diverse mutations of the AR have been reported in association with the phenotype of complete androgen insensitivity.
At the other end of the phenotypic spectrum are individuals in whom the male phenotype reflects a nearly normal level of virilization. Such affected individuals (often referred to as mild androgen insensitivity syndrome, MAIS) display normal phallic and scrotal development, but exhibit subtle signs of undervirilization, such as gynecomastia, poor beard growth, and infertility. Only a limited number of mutations in the AR have been described in such individuals. In all instances in which this has been examined, the mutations result in limited impairment of AR function in functional assays.
Between these two extremes of phenotypic development are individuals in whom substantial alterations of virilization are evident. Historically, these syndromes have been referred to using a variety of terms, including Reifenstein’s syndrome, partial androgen insensitivity syndrome (PAIS), and incomplete testicular feminization. In the most severely affected individuals, only minor amounts of virilization are evident. Such individuals may have a primarily female phenotype, but with only minor degrees of clitoromegaly. In individuals in whom a greater degree of AR function is preserved, significant virilization is present, but in the context of substantial defects of urogenital development, such as perineal hypospadias (the Reifenstein phenotype). Quigley et al. have employed a more detailed system with which to categorize patients with partial androgen insensitivity.
The genetic basis of androgen insensitivity
More than 500 different mutations have been described within the AR gene associated with androgen insensitivity, both within and adjacent to the coding region of the receptor protein.
The following summary groups the types of mutations that have been identified and presents generalizations regarding effects on receptor function and resulting phenotype for each of the categories. The locations of representative examples of the different mutation types are depicted in Fig. 20.2 .
Disruption of the primary amino acid sequence
Many mutations have been described that interrupt the AR open reading frame. These include deletions and insertions (both small and large scale) and mutations that result in changes in the splicing of the mRNA encoding the AR protein. As noted previously, the regions of the AR that are responsible for mediating the recognition of androgen-responsive DNA sequences and for the binding of ligand by the AR are located in the center and carboxyl terminal regions of the protein, respectively ( Fig. 20.2 ). For this reason, termination of the receptor within the open reading frame will result in the synthesis of an AR protein that is lacking part or all of one of these critical domains. In general, small insertions or deletions within the critical DBD and LBD that maintain the integrity of the open reading frame completely disrupt AR function.
Alterations of the DBD
In studies performed in genital skin fibroblasts from patients with clinical features, endocrine testing, and family history consistent with androgen insensitivity, approximately one-fifth displayed normal levels of qualitatively normal androgen ligand binding.
The AR genes from a number of such individuals have been analyzed. In virtually all instances, alterations of the structure of the DBD of the receptor have been identified. In one of the first series examining this class of AR defect, investigators found that when recreated in heterologous cells, the amino acid substitutions identified within the DBD of receptor protein had no effect on ligand binding, When examined in functional assays, however, these mutant receptors were impaired in their ability to bind to target DNA sequences and to stimulate a model reporter gene, findings consistent with results from a number of other laboratories. These findings are also relevant to alterations of the DBD caused by deletions and insertions and by alterations of mRNA splicing.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


