ANDROGEN DEPRIVATION THERAPY
Although the exact pathogenesis of BPH is not well defined, clearly aging and the presence of androgens are required for the development of this condition. Numerous laboratory and clinical studies have demonstrated that the prostate gland is an androgen-sensitive organ. If the androgenic stimulation to the prostate is eliminated, the prostatic adenoma decreases in size, outflow resistance through the prostatic urethra diminishes, and the ability to urinate improves.25,26 and 27 The predominant histologic effect is involution of the glandular epithelium of the prostate. To produce a state of androgen deprivation, the hypothalamic–pituitary–gonadal axis must be interrupted.
Under normal physiologic conditions, testicular androgen production is under the control of the hypothalamus.28 Neurons in the preoptic area of the hypothalamus secrete the decapeptide luteinizing hormone–releasing hormone (LHRH) (also called gonadotropin-releasing hormone [GnRH]) in a pulsatile fashion directly from their terminals into the hypophysioportal circulation. GnRH then interacts with its high-affinity receptor on the plasma membrane of the anterior pituitary cells to stimulate them to release the heterodimeric gonadotropins—follicle-stimulating hormone (FSH) and luteinizing hormone (LH). LH travels through the peripheral circulation to reach the testes, where it binds to its high-affinity receptor on the surface of the Leydig cells. These Leydig cells are stimulated to produce and secrete primarily testosterone (4-androsten-17β-ol-3-one), which travels in the bloodstream either in a free state or bound to sex hormone–binding globulin (SHBG) and albumin. At the target organ (the prostate gland), unbound testosterone binds to its high-affinity receptor within the prostatic cell, where the majority is converted to dihydrotestosterone (DHT) by the enzyme 5α-reductase.29 Inside the prostatic cell, the remaining testosterone and DHT bind to the same high-affinity androgen-receptor protein, enter the nucleus, and interact with specific DNA-binding sites. Androgen-dependent genes undergo transcription, and the resulting mRNA is translated into specific proteins. Overall, protein biosynthesis increases, leading to cellular hypertrophy and hyperplasia. Each of the three agents
most commonly used to inhibit testicular androgen production (GnRH agonists, antiandrogens, and 5α-reductase inhibitors) interferes with the hypothalamic–pituitary–gonadal axis (Fig. 121-1 and Fig. 121-2) to exert an effect on the enlarged prostate gland.
most commonly used to inhibit testicular androgen production (GnRH agonists, antiandrogens, and 5α-reductase inhibitors) interferes with the hypothalamic–pituitary–gonadal axis (Fig. 121-1 and Fig. 121-2) to exert an effect on the enlarged prostate gland.
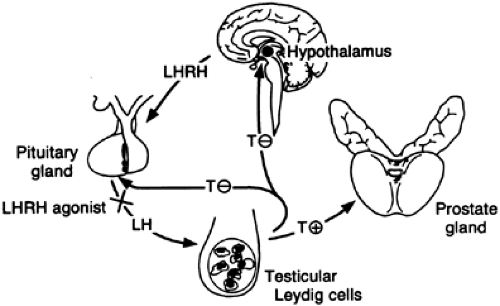 FIGURE 121-1. Hypothalamic–pituitary–gonadal axis. (LHRH, luteinizing hormone–releasing hormone; LH, luteinizing hormone; T, testosterone.) (From Oesterling JE. LHRH agonists. A nonsurgical treatment for benign prostatic hyperplasia. J Androl 1991; 12:381.) |
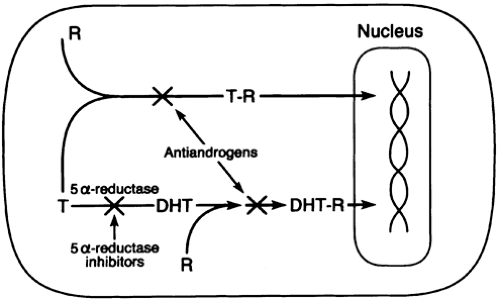 FIGURE 121-2. Diagram of prostate cell, showing the mechanism of testosterone (T) stimulation and its blockage by 5α-reductase inhibitors. (R, cytoplasmic receptor for androgens; T-R, testosterone receptor complex; DHT, dihydrotestosterone; DHT-R, dihydrotestosterone receptor complex.) (From Monda JM, Oesterling JE. Curr Probl Urol 1992; 2:100.) |
GONADOTROPIN HORMONE–RELEASING HORMONE AGONISTS
GnRH agonists bind to the GnRH receptor of the gonadotropic cells of the anterior pituitary gland. Normally GnRH is released in a pulsatile fashion by the hypothalamus and stimulates the production of LH. Paradoxically, administration of a GnRH agonist in a continuous fashion results in internalization of the GnRH receptor complex in the cells of the anterior pituitary. As the number of receptors on the plasma membrane decreases, the stimulation of these cells and subsequent LH release decrease.30,31 The administration of GnRH agonists is associated with an LH and testosterone flare, which rapidly reverses, and in several weeks castrate levels of serum testosterone and serum DHT are achieved.32 Like surgical castration, this medical castration decreases the size of the prostatic adenoma.
A number of clinical trials of GnRH agonists have been carried out. Initially, the use of the GnRH agonist buserelin was described for patients with BPH.33 Later nafarelin acetate was reported to be effective for patients treated for 6 months.34 In a subsequent uncontrolled study evaluating 17 patients in a 6-month trial of goserelin,35 many patients elected to continue the treatment for a year. Prostate size reached its nadir at 9 months with a mean decrease of 63%; this size decrease was accompanied by a significant improvement in peak urinary flow rates. No significant change was seen in the postvoid residual volume. Urinary symptoms decreased within the first month, with a mean decrease of 33% at 6 months. Six of the 17 patients experienced no improvement in urinary symptoms during the treatment period, and 3 months after the treatment was withdrawn, prostate volumes returned to 95% of the pretreatment volumes. Symptoms and urinary flow rates also worsened, indicating that the improvements could not be maintained in the absence of continuous treatment.
In most men the side effects associated with GnRH agonist therapy outweigh the benefits. Impotence and loss of libido occur in nearly all treated men, and hot flashes and gynecomastia develop in ˜50%. Other side effects include weight gain and a loss of energy. Treatment with GnRH agonists is also expensive (˜$5000 per year). For these reasons, and because of the availability of other treatment modalities, GnRH agonists are rarely used in the treatment of BPH.
ANTIANDROGENS
Androgen-receptor–blocking agents directly interfere with the ability of testosterone and DHT to bind to the androgen receptor within the prostate (see Fig. 121-2). The result is prostatic involution with normal serum testosterone levels.36 Thus, many of the side effects associated with GnRH agonists can be avoided.
Three antiandrogens for which clinical trials have been conducted are flutamide (Eulexin), bicalutamide (Casodex), and zanoterone. All three compounds are orally administered, non-steroidal compounds. Flutamide is metabolized in the liver to its active form, hydroxyflutamide. Both hydroxyflutamide and bicalutamide compete effectively with testosterone and DHT for the cytosolic androgen receptor.37,38 In rat models, bicalutamide has an affinity for the androgen receptor that is four times greater than flutamide.39 In addition, the serum half-life is one week, allowing for once-a-day dosing at a lower dosage than flutamide.37 The antiandrogens have no androgenic, estrogenic, antiestrogenic, progestational, or antiprogestational action; therefore, the serum testosterone level is not decreased, and fewer hormonally related side effects occur.40 Serum LH and testosterone levels may be elevated by flutamide because of the loss of central inhibition by the negative hypothalamic– pituitary–gonadal feedback loop. This effect is not seen with bicalutamide use because it is peripherally selective and has no effect on the hypothalamus. Both flutamide and bicalutamide have been approved by the Food and Drug Administration for use in the treatment of advanced prostate cancer.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


