Neoplastic/pathological hypercortisolism
ACTH-secreting neoplasm
Pituitary (Cushing disease)
Non-pituitary (ectopic ACTH)
Adrenal neoplastic disease
Adrenocortical adenoma
Adrenocortical carcinoma
Bilateral adrenal nodular disease
Primary pigmented micronodular hyperplasia
Primary bilateral macronodular hyperplasia
Nonneoplastic/physiological hypercortisolism
Phenotype similar to neoplastic hypercortisolism
Alcoholism and alcohol withdrawal
Chronic kidney disease stage 5
Depression/neuropsychiatric disease
Glucocorticoid resistance
Uncontrolled diabetes mellitus
Pregnancy
Phenotype not similar to neoplastic hypercortisolism
Starvation/malnutrition—anorexia nervosa
Critical illness
Aging
The purpose of this chapter is to review the medical disorders and physiological conditions that are known to be associated with chronic activation of the HPA axis and to provide a rational clinical and biochemical approach to help distinguish them from true pathological Cushing syndrome.
HPA Axis Physiology and its Potential Association with Common Disorders
The HPA axis exists primarily to generate a basal, circadian cortisol rhythm and to increase cortisol secretion in response to a wide variety of stimuli collectively termed, rather imprecisely, stress [1, 11, 12]. Attempts have been made to categorize stress into subtypes, such as psychological and physical [1]. These designations are particularly relevant to this chapter as we are emphasizing the differences and similarities between chronic stimuli to the HPA axis compared to the truly pathological, endogenous hypercortisolism characteristic of Cushing syndrome and usually due to an endocrine neoplasm .
Figure 1 gives an overview of the general structure of the HPA axis [11, 12]. CNS inputs to the hypothalamus from the circadian rhythm pathways [13] and stress pathways [1, 14–16] elicit an increase in the release of corticotrophin-releasing hormone (CRH) and/or arginine vasopressin into the hypophyseal portal veins which then stimulate the release of preformed ACTH as well as increase the transcription of the proopiomelanocortin (POMC) gene. ACTH is produced from POMC by post-translational processing and released into the systemic circulation. At the adrenal cortex, ACTH binds to and activates the melanocortin 2 (ACTH) receptor leading to an activation of the cAMP-steroidogenic-acute regulatory (StAR) protein cascade which increases steroidogenesis within the adrenal zona fasciculata cell [17, 18]. Cortisol, released into the blood from the adrenal cortex, binds >90 % to plasma proteins (primarily CBG at physiological cortisol concentrations) and circulates throughout the body. At the hypothalamus and anterior pituitary, free (biologically active) cortisol exerts a negative feedback effect via both the glucocorticoid (GR) and mineralocorticoid (MR) receptors [19, 20]. Previous chapters in this book have elaborated in great detail on the mechanisms of action of circulating cortisol. Suffice it to say here that target tissues for glucocorticoids have an elaborate enzymatic system for protecting the MR from the actions of cortisol at physiological concentrations [21]. This is particularly relevant to this chapter as many of the effects of cortisol excess, such as increased sodium retention and potassium excretion in the kidney, are due to the saturation of these protective mechanisms and the binding of cortisol to the MR [22–24].
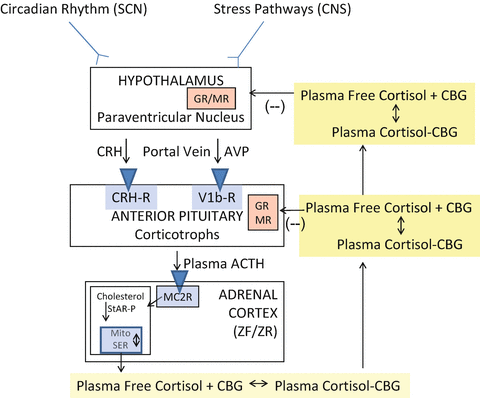
Fig. 1
General organization of the hypothalamic–pituitary–adrenal axis. Suprachiasmatic nucleus (SCN); central nervous system (CNS); corticotrophin-releasing hormone (CRH); arginine vasopressin (AVP); adrenocorticotropic hormone (ACTH); zona fasciculata (ZF); zona reticularis (ZR); melanocortin 2 receptor (MC2R); StAR-P (steroidogenic acute regulatory protein); mitochondria (Mito); smooth endoplasmic reticulum (SER); corticosteroid-binding globulin (CBG, also known as cortisol-binding globulin); Glucocorticoid receptor (GR); mineralocorticoid (MR) receptors. From [11]
A few of the points above deserve elaboration. One of the hallmarks of the diurnal mammal is the circadian rhythm of the HPA axis in which cortisol peaks between 0600 and 0800 h and is at its nadir around midnight or a little later [13, 25]. The HPA axis also exhibits ultradian rhythmicity which may account for some of the “noise” in the assessment of morning cortisol concentrations in plasma [26, 27]. As described later, one of the earliest and most consistent changes in the HPA axis in patients with Cushing syndrome is an increase in the late-night nadir in cortisol [26, 28–32]. As you will see, this is exploited in the diagnosis of Cushing syndrome.
As mentioned above, there are many neuropsychiatric situations in which the normal circadian rhythm of the HPA axis is disrupted [33]. Although chronic increases in HPA activity due to neuropsychiatric disorders are described below, it is also important to point out that patients with these disorders can exhibit an augmented response to stress despite the fact that basal cortisol secretion can be normal [33]. In addition, the termination of the HPA axis response to stimuli in these conditions can also be delayed leading to even more exposure to increased glucocorticoid activity [33]. Therefore, just because a basal cortisol level is within the reference range does not mean the person has not been repetitively exposed to increased cortisol levels during the stresses of everyday life.
Another interesting characteristic of the HPA axis that is becoming of great physiological and psychoneuroendocrinological interest is the increase in cortisol secretion that occurs upon awakening—the cortisol awakening response (CAR )—that is superimposed on the circadian rhythm described above [34–41]. It has been proposed that a change in the CAR is an indication of alterations in arousal, anticipation of the day’s events, gender, health status, and the perception of stress. The CAR can easily be assessed by measuring salivary cortisol immediately upon awakening and then at set times thereafter [37]. Of great interest is the possibility that the CAR may reflect changes in neurological function especially in the hippocampus and associated limbic structures [40]. Since endogenous depression is one of the common maladies we will focus on as a state of hypercortisolism, it is interesting that the CAR is increased during episodes of major depression [38].
In summary, it is now clear that exposure to increased cortisol may be quite common in everyday life particularly in people with neuropsychiatric and other medical conditions.
Diagnostic Tests for Endogenous Hypercortisolism
A recent Endocrine Society Guideline has proposed three screening tests for Cushing syndrome that exploit different aspects of the disruption of normal physiology [3, 42]. They are as follows: the failure to achieve the normal nadir in late-night cortisol most commonly performed by the measurement of an increased late-night (typically at bedtime) salivary cortisol; the failure to suppress morning plasma cortisol after an overnight dexamethasone suppression; and an increase in the excretion of free cortisol in the urine. We have extensively reviewed these “first line” tests previously [7, 8, 11, 12, 32, 43] and will only briefly describe them below.
Late-Night Cortisol : It was observed decades ago that patients with severe endogenous Cushing syndrome of any etiology show a disrupted cortisol circadian rhythm [44–47]. More recently, it was demonstrated that patients with milder forms of Cushing syndrome have increased midnight plasma cortisol and that this measurement when done properly can be used to diagnose and rule out Cushing syndrome with accuracy [48, 49]. Considering the challenge of obtaining stress-free late-night blood samples in patients, a major advance came with the development and widespread clinical use of the measurement of late-night salivary cortisol (LNSC) as a surrogate for plasma free cortisol [12, 50, 51]. This test typically has a sensitivity and specificity for endogenous hypercortisolism of >90–95 %, although there are some exceptions and methodological caveats that must always be kept in mind [51, 52].
Low-Dose Dexamethasone Suppression Test : The physiological concept exploited in this test is that ACTH-secreting neoplasms (and obviously ACTH-independent adrenal neoplasms) have attenuated sensitivity to cortisol negative feedback [6, 7, 53]. The failure to fully suppress morning cortisol after an overnight (usually 1 mg) dose of dexamethasone (i.e., cortisol >1.8 μg/dL [50 nmol/L]) has a sensitivity of 95 % in patients with neoplastic Cushing syndrome; however, there are many factors that contribute to false positive results yielding a diagnostic specificity of 85–90 %. Most common causes of misleading results are concomitant use of drugs which accelerate or impair dexamethasone metabolic clearance and the use of estrogen therapy which increases corticosteroid-binding protein (CBG), the major binding protein for cortisol.
Urine Free Cortisol ( UFC ): The physiological concept is that an increase in the filtered load of free cortisol in the kidney will be reflected in an increase in 24-h UFC excretion. However, this assumption, which was based on old immunoassay data, has recently been questioned [54, 55]. The increasing use of the highly specific measurement of UFC using LC-MS/MS could theoretically resolve some problems related to this test [56]. This may not be true because of the theoretical advantage of measuring cortisol metabolites in the urine [54]. Although measurement of UFC has a sensitivity of only 75 % for the detection of neoplastic Cushing syndrome, marked elevations of urine cortisol (>3–4 times the ULN) are virtually diagnostic of pathological Cushing syndrome [3]. Nonetheless, because of its very poor sensitivity we do not recommend UFC as a first line test in the evaluation of suspected hypercortisolism .
Physiological (Nonneoplastic) Hypercortisolism
There are a variety of circumstances not attributable to an ACTH- or cortisol-secreting neoplasm in which cortisol secretion is chronically increased. This section will discuss some of these rather common situations and will express our opinion that these varied states of mild to severe cortisol excess should not be lumped together, but rather discussed on their own merits. These often subtle situations activate the HPA axis primarily through neural pathways with input to the parvocellular paraventricular nuclei in the hypothalamus (see Fig. 1). In reality as you will see, one mechanism that is a recurring theme in many of these situations is a decrease in sensitivity to glucocorticoid negative feedback . This can lead to mild increases in cortisol levels more equivalent to subclinical Cushing syndrome. However, it is very important to again emphasize that small increases in cortisol can summate over time to considerable glucocorticoid exposure [57].
Alcohol-Induced Hypercortisolism : It has been known for many years that alcohol intake increases cortisol secretion acutely and that actively drinking alcoholics have increased indices of cortisol secretion compared to controls [58, 59]. The mechanism for this increase is thought to be centrally mediated due to increases in CRH and ACTH [60]. In the late 1970s, investigators began to recognize the presence of signs and symptoms of Cushing syndrome in alcoholic patients with resolution of the clinical features and biochemical abnormalities of cortisol excess within 1–2 months after abstinence from alcohol [61, 62].
Experimental studies suggest that the principal stimulus of alcohol-induced cortisol secretion is centrally mediated through hypothalamic CRH [63–65]. Messenger RNA for CRH is increased in the paraventricular nucleus of rats after alcohol administration, and in addition, CRH receptor antagonists abolish the ability of alcohol to stimulate the HPA axis [63, 64]. Alcohol administration does not stimulate the HPA axis in rats after hypophysectomy or suppression of hypothalamic activity with the administration of morphine and pentobarbital [66]. Alcohol-induced increases in vasopressin secretion may also be a factor, since hypothalamic vasopressin of parvo- and magnocellular origin augments the ACTH response to CRH. Removal of endogenous AVP diminishes the alcohol-evoked ACTH secretion in both sham-operated and paraventricular nucleus-lesioned rats [63, 64]. Nonetheless, Cobb et al. have shown that steroid production from isolated perfused rat adrenal glands increases after adding ethanol in the absence of ACTH and that the effect is not enhanced by ACTH administration [67]. However, Elias et al. showed that a large alcohol bolus did not cause an increase in plasma cortisol or ACTH levels and failed to potentiate the effect of exogenous ACTH on cortisol secretion in either alcoholic subjects or normal human subjects [68]. Wand et al. reported increased ACTH levels at 1400 h with concurrently normal cortisol values in 31 actively drinking alcoholics [60]. The normal cortisol in the presence of increased plasma ACTH suggests centrally mediated HPA axis hyperactivity . Withdrawal from alcohol in chronic alcoholics also causes increases in ACTH and cortisol and normalization of HPA axis function may require a few weeks after alcohol cessation [69].
Altered peripheral metabolism of cortisol (particularly in the liver) may contribute to hypercortisolism in these patients. Lamberts et al. reported a patient with alcoholism with clinical Cushing syndrome and suppressed plasma ACTH [70]. They demonstrated a prolonged half-life of cortisol; however, if negative feedback system functioned properly, one would expect cortisol levels to eventually return to normal unless stimulatory input to the hypothalamus was increased. Moreover, some investigators have failed to detect a causal relationship between the impairment of liver function per se and serum cortisol levels [71]. Nonetheless, it is not uncommon for patients with alcohol-induced hypercortisolism to have abnormal liver function studies. Consequently, the presence of persistent liver function abnormalities —particularly if the AST is much greater than the ALT—should raise concern about the possibility of excessive alcohol consumption [72].
Another potential factor in hypercortisolism from excessive alcohol consumption might be interference of the binding of cortisol to plasma proteins with possibly excessive free cortisol concentrations . One study showed a positive correlation between blood alcohol level and the percentage of free plasma cortisol [73]. There was a shift of the fraction of cortisol bound to cortisol-binding globulin to the albumin-bound and unbound fractions. They speculated that there may be an intracellular hypoglucocorticoid state which gives rise to stimulation of the HPA axis in patients with normal cortisol negative feedback control. Since alcohol-induced hypercortisolism is not commonly appreciated in all alcoholic subjects, there may also be genetic influences that have an impact on the effect of alcohol in the HPA axis.
Biochemical studies in patients with alcohol-induced hypercortisolism have shown normal, increased, and occasionally even decreased concentrations of plasma ACTH [60, 70]. Diurnal rhythm is usually absent or attenuated and urinary measurements of corticosteroids are often increased. Overnight dexamethasone suppression testing yields abnormal results in the majority of patients with alcohol-induced hypercortisolism and the ACTH response to CRH is either normal or blunted [70, 74–76]. The dexamethasone-CRH test has been shown to be abnormal in alcohol-induced hypercortisolism and cannot be used to discriminate this from pathological Cushing syndrome [77]. On the other hand, the ACTH response to desmopressin acetate (DDAVP) appears to be absent in alcohol-induced Cushing syndrome (like normal subjects) in contrast to patients with Cushing disease [78]. Finally, using salivary cortisol measurements , it has been shown that alcohol intoxication activates the basal HPA axis but appears to blunt the stress response [79, 80].
In our experience, alcohol-induced hypercortisolism can be difficult to distinguish clinically or biochemically from patients with pathological Cushing syndrome. This is a particular problem if patients are not forthright about the magnitude of their alcohol consumption. The majority of patients have normal or increased plasma ACTH, so the central effects of alcohol in the HPA axis seems to predominate. Nonetheless, we have, like others, observed adrenal nodular disease in patients with alcohol-induced hypercortisolism and this disorder may actually present as an incidental adrenal nodule in some patients [81].
In summary, the etiology of alcohol-induced hypercortisolism is uncertain and, in fact, there may be several interrelated causes that can vary in significance in individual patients. It is clear that it can resolve within a few weeks of alcohol cessation [70, 76], although there may be long-lasting effects on stress responsivity of the HPA axis [80]. Clinical and biochemical features are often indistinguishable from patients with true pathological Cushing syndrome. A high index of suspicion may be necessary to make an accurate diagnosis, and secondary testing with the DDAVP stimulation test would seem to be the best diagnostic option.
Starvation : One of the most important adaptive responses to starvation is activation of the HPA axis to liberate energy stores and stimulate gluconeogenesis in order to maintain plasma glucose in the normal range. Many studies have demonstrated that patients with eating disorders (specifically anorexia nervosa) have activation of the HPA axis with varying degrees of hypercortisolism usually with normal plasma ACTH [46, 82]. Patients with anorexia nervosa have altered HPA dynamics with increases in urinary cortisol excretion and late-night salivary cortisol as well as abnormal dexamethasone suppression [83]. Typically, patients with anorexia nervosa have an attenuated ACTH response to CRH most likely due to the inhibitory feedback of cortisol on the anterior pituitary [46]. The dexamethasone-CRH test may also be abnormal in patients with anorexia [84]. Similar to normal subjects, DDAVP does not stimulate ACTH and, hence, cortisol in patients with anorexia. Furthermore, DDAVP can enhance ACTH and cortisol release after CRH in normal subjects but not in patients with anorexia nervosa [85]. All these findings point to an attenuation of the pituitary corticotroph response to endogenous stimuli.
Anorexia nervosa-induced hypercortisolism correlates with the severity of bone loss in women and has also been shown to be associated with the hypothalamic amenorrhea [86, 87]. In addition, increased bone marrow fat related to cortisol excess has also been reported in patients with anorexia nervosa [83]. It seems likely that other starvation-equivalent disorders may be associated with hypercortisolism and have significant clinical manifestations. For example, patients with prolonged stay in the intensive care unit have been known to have significant myopathy and a catabolic state mediated, in part, by hypercortisolism [88]. Increases in cortisol have also been observed in a study of normal weight women undergoing low-calorie dieting, and increased morning cortisol levels have been demonstrated in women within 6–12 months following bariatric surgery [89]. It seems possible that the chronic wasting and catabolic state seen in many serious chronic conditions (malignancies, cardiac, neurological, or infectious disorders) may be related, in part, to HPA axis activation and endogenous hypercortisolism. It is appreciated that patients with very severe hypercortisolism (usually ectopic ACTH secretion) may present with significant weight loss, edema, and myopathy [90]. In contrast to patients with starvation-induced hypercortisolism, patients with pathological Cushing syndrome almost always have insulin resistance and/or hypertension [5, 7].
In summary, central activation of the HPA axis with endogenous hypercortisolism is a common adaptive response to starvation. These disorders rarely cause any diagnostic confusion from patients with pathological Cushing syndrome. However, it should be pointed out that there are a few reports of anorexia nervosa as the initial clinical feature in patients with pathological Cushing syndrome as a reflection of the remarkably varied neuropsychiatric impact on the brain from cortisol excess [91].
Depression/Neuropsychiatric Disorders : There are a number of neuropsychiatric disorders that can increase or decrease the activity of the HPA axis [33]. It is not possible to go into great detail here. Rather, Table 2 provides a brief summary.
Major depression (melancholic, bipolar, or psychotic): |
Decreased sensitivity to glucocorticoid negative feedback |
Association with early life stress |
Anxiety/panic disorder |
Obsessive-compulsive disorder |
Schizophrenia: |
Decreased sensitivity to glucocorticoid negative feedback |
Autism spectrum disorder: |
Increased stress response |
There is a considerable history of the study of the HPA axis in depression [33]. It is generally accepted that most forms of major depression exhibit increased HPA axis activity. New data suggest that changes in glucocorticoid receptor, and possibly mineralocorticoid receptor sensitivity, lead to resistance to cortisol negative feedback inhibition [92]. Interestingly, successful pharmacotherapy seems to normalize HPA axis activity and the lack of effectiveness of therapy correlates with a persistence of HPA axis hyperactivity [33]. It is well known that patients with major depression (e.g., psychotic depression) often have abnormal low dose dexamethasone suppression as well as elevations of LNSC and UFC. In fact, mental health specialists have utilized not only the low dexamethasone suppression test but also the dexamethasone-CRH test to characterize these disorders and the response to therapy. Since neoplastic/pathological Cushing syndrome is often complicated by significant neuropsychiatric illness, the differentiation from HPA axis activation due to severe forms of depression is challenging. The DDAVP test has not been extensively studied in depressive illness, and variable ACTH and cortisol responses have been reported.
Aging: Healthy aging is associated with an increase in late-night salivary cortisol levels [57]. Again, this increase is not into the pathological range. However, the doubling of late-night salivary cortisol with healthy aging represents a doubling of exposure to bioactive (free) cortisol which is a significant glucocorticoid exposure integrated over time. In fact, this increase correlates with a decrease in bone mineral density—a potential surrogate for glucocorticoid exposure over time [57].
Like depression described above, the mechanism of the increase in HPA axis activity with healthy aging is associated with a decrease in sensitivity to cortisol negative feedback [93–95]. Also of interest is that there appears to be an interaction with aging and the development of depression in terms of the increase in HPA axis activity [96].
Chronic Kidney Disease: CKD and end-stage renal failure have long been known to be associated with dysregulated cortisol excess with abnormal dexamethasone suppression [97–99]. Recently, patients with end-stage renal failure receiving hemodialysis have been shown to have disrupted circadian rhythm and increased late-afternoon to late-night cortisol [25]. The mechanism for the increase in cortisol in CKD5 appears not to be the result of decreased renal clearance of cortisol since plasma ACTH is increased in these patients (Fig. 2). If this was a cortisol clearance defect, ACTH should be suppressed due to negative feedback as occurs with sepsis as described below [100, 101]. It seems clear from these findings that patients with end-stage renal disease have an activation of their HPA axis, presumably of hypothalamic origin. It has also been suggested that this activity correlates with increases in C-reactive protein suggesting that a heightened inflammatory state may be an etiology [25]. The secondary tests outlined below (Dex-CRH or DDAVP) have not been studied in patients with severe CKD.
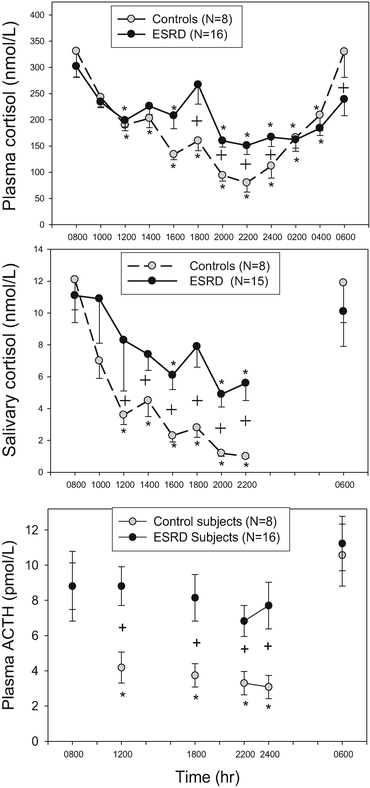
Fig. 2
Circadian rhythm of plasma cortisol (top), salivary cortisol (middle), and plasma ACTH (bottom) in control subjects compared to ESRD subjects. From [25]
Other Common Disorders with Subtle Increases in Cortisol: As discussed above, there are many disorders in which cortisol levels are not increased above the reference range but, rather, demonstrate subtle increases that, when integrated over time, can result in significant glucocorticoid exposure. Examples of these are hypertension [102] and type 2 diabetes mellitus (T2DM) [103].
Multiple sclerosis represents an interesting disease state with activation of the HPA axis [104]. At first, one might think this is due to the increased inflammatory state and cytokine stimulation of the HPA axis. It seems, rather, that this is due to a disruption of normal hypothalamic control directly due to brain lesions [104]. Another common state one might expect a dramatic activation of the HPA axis is obstructive sleep apnea (OSA ) due to frequent arousals [39] and hypopnea leading to hypoxia [105]. Interestingly, patients with OSA seem remarkably resistant to significant activation of the HPA axis [105], although they may have an increased CAR [39].
Pregnancy: The increase in serum-free and salivary cortisol that occurs during pregnancy is well documented [106–108]. It is also well known that serum CBG increases during pregnancy due to the effect of estrogen on hepatic production [106]. The increase in total cortisol that results would not be expected to dramatically increase free (biologically active) cortisol concentrations in the blood. However, the increase in free (bioactive) plasma cortisol that has been demonstrated is due to the increase in plasma ACTH as pregnancy progresses [109]. Several mechanisms have been proposed for the increase in plasma ACTH including the secretion of CRH from the placenta, the increase in progesterone that can act as a glucocorticoid antagonist, a decrease in glucocorticoid negative feedback sensitivity, and production of ACTH from the placenta [106]. It is important to point out that CRH-binding protein also increases during pregnancy which mitigates some of the effect of placental CRH on the maternal pituitary gland [110–112]. In addition, there does appear to be a decrease in corticotroph sensitivity to unbound plasma CRH in late pregnancy [112]. The increase in bioactive (free) cortisol during pregnancy may help to sustain maternal gluconeogenesis to maintain delivery of glucose to the fetus.
Critical Illness: In the classic differential diagnosis of Cushing syndrome, ACTH independence is either due to adrenal autonomy in endogenous disease or due to glucocorticoid therapy in exogenous disease. Any state in which cortisol is increased and ACTH is suppressed can be categorized as ACTH independent. The case of critical illness usually due to sepsis is unusual in this regard and has been a source of confusion for decades [100, 113]. It is acknowledged that early in the development of sepsis and often before the patient has been admitted to the ICU, ACTH does increase (transiently) driving the adrenal gland to increase cortisol production [114]. Therefore, the initial stimulus to the axis seems to be of hypothalamic and pituitary origin. Thereafter, there is a sustained increase in plasma cortisol in the face of decreased plasma ACTH even though the stimulus (i.e., sepsis and hypotension) is still present [101]. This sustained increase in plasma cortisol is thought to be due to a decrease in the metabolic clearance rate of cortisol [100, 101]. Therefore, the final rate of ACTH secretion from the anterior pituitary gland is a balance between stimulatory input from stress pathways and inhibitory cortisol negative feedback. The decrease in CBG concentration often found in septic ICU patients may lead to a state of bioactive cortisol excess [115, 116]. Ironically, the prolonged suppression of ACTH secretion combined with decreased perfusion pressure in patients in the ICU for extended periods of time may eventually result in the risk for the development of adrenal insufficiency [100]. These phenomena do not seem to be unique to severe sepsis as trauma and cirrhotic patients also appear to exhibit some of the same characteristics [117, 118].
Type 2 Diabetes , Insulin Resistance , and the Metabolic Syndrome : One of the ongoing controversies in endocrinology is whether the frequency of pathological Cushing syndrome is more common in patients with T2DM. Although initial reports suggest a higher frequency of Cushing syndrome in patients with T2DM [119, 120], subsequent studies have suggested that only T2DM patients with pathognomonic features of endogenous hypercortisolism should be evaluated for a neoplastic cause [121–123]. It is well known that glucocorticoid therapy causes insulin resistance. A related question is whether subtle disruptions of the HPA axis can contribute to the development of the metabolic syndrome and insulin resistance. Increased late-night salivary cortisol concentrations have been found in poorly controlled patients with T2DM [124]. However, a minimal association of glycemic fluctuations with salivary cortisol excursions has subsequently been described [125]. A fascinating study of a Namibian ethnic group during urbanization found an association of salivary cortisol increases with a disruption of glucose homeostasis [126]. Finally, the concept of “tissue-specific Cushing syndrome ” has been suggested in patients with obesity, the metabolic syndrome, and insulin resistance [127, 128]. Specifically, increased adipose expression of 11BHSD1 theoretically generating increased tissue cortisol levels has been proposed [129]. This raises the possibility that medical therapy directed at 11BHSD1 may be useful in the treatment of obesity. In summary, there is moderate evidence that subtle alterations in HPA axis activity can contribute to the development of insulin resistance. In addition, selected patients with clear and specific features of Cushing syndrome and poorly controlled T2DM should be screened for neoplastic causes of endogenous hypercortisolism .
Glucocorticoid Resistance: Glucocorticoid (or primary cortisol) resistance is reviewed in detail elsewhere in this book and therefore will only be discussed in brief here. This is typically a familial receptor-mediated disorder that presents with increased androgen and cortisol production in an otherwise healthy individual [130]. Since the index cases are usually diagnosed in adulthood, the cortisol resistance is partial and accompanied by compensatory increases in circulating pituitary ACTH, and cortisol with excessive secretion of adrenal androgens and adrenal steroid biosynthesis intermediates with salt-retaining activity (e.g., deoxycorticosterone). The clinical manifestations of glucocorticoid resistance include chronic fatigue (possibly due to the result of glucocorticoid deficiency in the central nervous system) and various degrees of hypertension with or without hypokalemic alkalosis and hyperandrogenism [131]. These patients do not have the catabolic features of hypercortisolism such as cutaneous wasting, abdominal striae, myopathy, and low bone density. Nonetheless, the excessive adrenal mineralocorticoid secretion, hypokalemia, and hypertension with hypercortisolemia may be confused with pathological Cushing syndrome. In women, hyperandrogenism can result in hirsutism, menstrual irregularities, and oligomenorrhea with decreased fertility and often mimics the polycystic ovary syndrome [130]. In men, glucocorticoid resistance may lead to infertility and, in children, to precocious puberty [132]. The peripheral tissues are relatively insensitive to cortisol but they maintain sensitivity to androgens and mineralocorticoids. Normal circadian rhythm is maintained in glucocorticoid resistance; however, since the cortisol levels are reset at a higher concentration, late-night salivary cortisol levels will be elevated.
The hypertension of glucocorticoid resistance is volume dependent and associated with low plasma renin activity and sometimes hypokalemic metabolic alkalosis [133]. High circulating concentrations of deoxycorticosterone and cortisol mediate the hypertension. The high cortisol levels overwhelm the intrarenal metabolic clearance of cortisol to cortisone by 11β-hydroxysteroid dehydrogenase type 2 and participate in the generation of hypertension by binding to the mineralocorticoid receptor.
The inheritance patterns are variable, and both autosomal dominant and recessive inheritance have been described [133]. Generally, in the dominant syndromes, the mutant glucocorticoid receptor interferes with the function of the normal receptor causing a so-called dominant-negative effect . In the recessive syndromes, the normal receptor tends to rescue the mutant receptor so that heterozygotes are clinically normal.
Clinical Discrimination of Physiological and Pathological Hypercortisolism
The most important way to separat e patients with pathological hypercortisolism from those with a physiological cause is to take a detailed history and perform a good physical examination. Chronic alcoholism, major depressive illness, and use of opioids are often the most difficult historical landmarks to document in patients with hypercortisolism. Many patients with chronic alcohol abuse may underestimate or underreport their alcohol abuse and withhold significant information about such things including binge drinking, alcohol withdrawal syndrome, and arrest for driving while intoxicated [60]. At times, a high index of suspicion is needed to elicit an accurate history and sometimes clues such as persistent increases in liver function tests (particularly when the AST is much greater than the ALT) may provide clues for possible heavy alcohol consumption. Although opioids actually suppress HPA axis function (mediated by hypothalamic CRH suppression), there is an abrupt recovery and actually a hyperactive HPA axis response once the opioid is discontinued or its effect wanes [134]. Consequently, the evaluation of the pituitary–adrenal function in patients taking narcotics may be especially difficult.
As described above, some neuropsychiatric disorders (particularly major depressive illness) may activate the HPA axis and cause dysregulated cortisol hypersecretion. Since neuropsychiatric and neurocognitive dysfunctions are common manifestations of patients with pathological hypercortisolism, the presence of significant melancholia in a patient with hypercortisolism provides special challenges. Mental health specialists may need to be consulted to help with characterization of neuropsychiatric disorders. Nonetheless, the broad spectrum of neuropsychiatric disorders including obsessive-compulsive disorder, bipolar disorder, schizophrenia, and major depressive illness has all been reported in patients with pathological Cushing syndrome [135].
Poorly controlled diabetes has also been associated with hypercortisolism, but it is not clear what level of hyperglycemia actually activates the HPA axis and may cause diagnostic confusion [124, 136]. Generally, when pathological hypercortisolism causes poorly controlled diabetes mellitus, the clinical and biochemical diagnosis is usually straightforward. However, in many patients it may be necessary to use aggressive hypoglycemic pharmacotherapy to improve glycemic control before inaugurating diagnostic tests for possible Cushing syndrome. Many of the other disorders associated with hypercortisolism such as pregnancy, severe CKD, and chronic intense exercise can usually be easily established with a simple history and routine laboratory tests.
Related posts:
 and Treatment of Aggressive Corticotroph Pituitary Tumors
Regulation of Body Composition and Metabolism
Regulation of Neurocognitive and Neuropsychiatric Function
Strategies for Localization of ACTH-Secreting Tumors
and Treatment of Aggressive Corticotroph Pituitary Tumors
Regulation of Body Composition and Metabolism
Regulation of Neurocognitive and Neuropsychiatric Function
Strategies for Localization of ACTH-Secreting Tumors
 Metabolism as a Regulator of the Tissue-Specific Glucocorticoid Action
Therapies in Cushing’s Syndrome
Metabolism as a Regulator of the Tissue-Specific Glucocorticoid Action
Therapies in Cushing’s Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


