Fig. 1
Comparison of liver cancer incidence in 2012 (Acknowledgment: adapted from IARC Globocan 2012) (a) and HBV chronic carriage prevalence in 2006 (Acknowledgment: adapted from Center for Disease Control and Prevention 2012) (b). Note that with the exception of Egypt due to a very high rate of HCV infection, liver cancer incidence in 2012 is higher in regions where HBV prevalence was high in 2006. See text for details in Mongolia and Laos
Indeed, HBV endemic has been linked to HCC, and it is well known from the prospective work of Beasley from 22,707 Taiwanese patients followed for 11.25 years [1] that there is an excess risk of dying of liver cancer among hepatitis B surface antigen (HBsAg) carriers compared to non-carrier (relative risk = 217). Furthermore, it has been very well sustained that the risk of chronic HBV carriage is the highest (up to 90 %) when the patient is infected at birth or during early childhood, whereas this risk is more in the range of 5–10 % when infected during adult life. Therefore, offspring when infected at birth from an HBV-infected mother are more susceptible to developing HCC in the future. Of particular interest in Taiwan is the fact that when a mass vaccination program against HBV to immunize newborn infants was implemented in 1984, the hepatitis B carrier rate in children covered by the program decreased from 15 to <1 % 20 years after. Most importantly, hepatocellular carcinoma in the vaccinees was also found to decrease in parallel. This is the first example that a human cancer might be prevented by a vaccination against its virally associated etiology.
This HBV transmission prevention by vaccination will profit to young new generation. Today, considering the still high number of chronic HBV carriers (n = 240,000,000), and due to the poor prognosis of HCC, deciphering the mechanism of HBV-associated cancer liver development is mandatory. If HBV infection by itself is an important risk factor, complex interactions between HBV viral elements and multiple host and environmental factors influence HCC. For example, hepatitis C virus (HCV) (see chapter “Hepatitis virus scientific background: Epidemiology and mechanism of carcinogenesis of hepatitis C virus (HCV)”) or hepatitis delta virus (HDV) [2] coinfections; exposure to aflatoxin B1 (AFB1) [3, 4]; alcohol and tobacco consumption; and metabolic diseases, such as nonalcoholic steatosis hepatitis (NASH) [5] and diabetes [6], might also be taken into consideration for cofactors or comorbidities implicated in liver carcinogenesis.
2 The HBV Natural History May Induce Chronic Inflammatory Infection
Hepatitis B virus primary infection may be asymptomatic or may cause an acute illness with symptoms including jaundice, dark urine, fatigue, and digestive-associated complaints. Hepatitis B prevalence is highest in sub-Saharan Africa and East Asia (≥8 %, see Fig. 1b), where transmission from generation to generation occurs. High rates of chronic infections are also found in the Amazon and the southern parts of Eastern and Central Europe. In the Middle East and the Indian subcontinent, an estimated 2–5 % of the general population is chronically infected, whereas <1 % of the population in Western Europe and North America is chronically infected (Fig. 1b). What had been noted for a long time is that the liver cancer incidence is higher in areas with high HBV prevalence (see Fig. 1).
In individuals chronically infected by HBV, the need for efficient antiviral therapy remains clear when a viral replication is observed to control the risk of progression and delay liver transplantation, which represents the only end-stage treatment. Indeed, patients having advanced chronic hepatitis B (CHB) can now be successfully treated using nucleos(t)ide analogs (NA) or pegylated interferon (PEG-IFN). Therefore, beside vaccination, prevention of the progression of the disease to cirrhosis and liver decompensation, leading to end-stage liver disease and/or to hepatocellular carcinoma, by inhibiting viral replication, represents a logical approach to improve survival.
During chronic infection, liver regeneration (renewal of the pool of cell) and architecture reconstruction (due to stimulation of stellate cells) will lead to activation of cell cycle and fibrogenesis, respectively (Fig. 2). Collagen scar tissue, in response to liver injury, may lead to nodular reconstruction pattern of fibrosis, progressively interrupting the normal communication between portal space and subhepatic venous circulation leading to liver cirrhosis. This advanced state of nodular fibrosis is often present in the West countries before the emergence of HCC and corresponds to a risk factor by itself. In a Western series of 30 patients without cirrhosis, all patients had some non-tumorous pathological changes such as iron overload and large cell dysplasia in their liver [7]. In contrast, in developing countries, cirrhosis is typically overshadowed by the signs of the tumor, unfortunately when it has reached an advanced stage. Indeed, analyzing 14 trials (six Asian and eight non-Asian) [8], Hsu and coworkers found that median survival rate of HCC reflecting natural history was 3.57 ± 1.88 month in Asian trials versus 5.96 ± 1.46 in non-Asian trials.
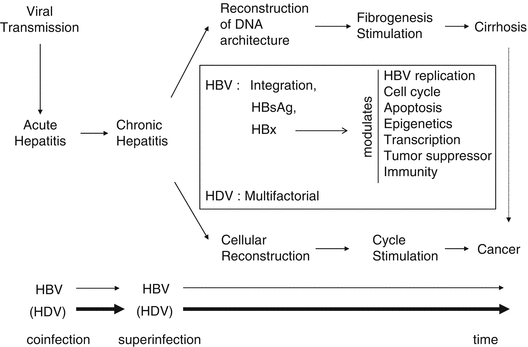
Fig. 2
After HBV transmission, infection leads to chronicity especially in neonates (90 %) and in adults (5–10 %). Due to liver inflammation, both liver fibrosis and cell regeneration are stimulated leading to cirrhosis and cell proliferation. Cirrhosis is by itself a precancerous state. In the square are simplified possible viral-induced mechanisms of hepatocarcinogenesis
3 The Hepatitis B Virus
Both HBV and hepatitis delta virus (HDV) use heparan sulfate membrane-anchored molecules and the Na taurocholate cotransporting polypeptide (NTCP) receptor to infect hepatocytes [9, 10]; L-HBsAg protein, coded by HBV PreS1/PreS2/S gene, is required for infection. The replication of the two viral genomes is fully independent in the nucleus of the cell. HBV DNA genome replicates through an RNA intermediate depending on its own viral polymerase having a reverse transcriptase activity, which is the step that will be inhibited by nucleoside/nucleotide analogs. Even under this therapy, the HBV DNA genome will persist as an episome in the nucleus of the infected cell. HBV genome can also integrate into the cellular genome (see below).
During viral replication, the intracytoplasmic reverse transcription step of the viral polymerase introduces errors that are not corrected due to the lack of a 3′→5′ exonuclease activity leading to mutations. In a chronically infected patient, more than 10E11 viral particles are produced per day, and viral polymerase introduces non-corrected errors every 10E4 to 10E5 nucleotides. Viral load and punctual mutations might also be involved in HCC. For example, the REVEAL study indicated clearly that the serum HBV DNA level is significantly and independently associated with incidence of HCC. In addition, viral genetic features such as core promotor mutations A1762T/G1764A mutant or precore G1896A were documented as predictor of HCC risk [11].
Eight different genotypes, A to H, have been described. Recently, a ninth “genotype” evidenced in North-West China, India, Laos, and Vietnam and tentatively termed “I” was suggested, although it is still subject to debate, as being a recombinant strain having a genotype C backbone. Finally, a tenth genotype provisionally assigned to genotype “J” was proposed for a Japanese patient’s HBV isolate.
Genotype-specific HBV genomes have sizes ranging from 3,182 nt (HBV/D) to 3,248 nt (HBV/G). This has consequences on the size of the viral proteins. Genotypes and subgenotypes might also be associated to specific geographic distributions reflecting ancient evolution. However, associated to human migrations, this picture may evolve. Furthermore, in some specific area, the recombinant strain represents the dominant variant such as the HBV/CD recombinant in Tibet [12]. Indeed, high endemy and coexistence of different genotypes in borders will favor a recombination process such as the recently described HBV/DE recombinant in Niger and India [13, 14].
Whether or not a specific viral genotype/subgenotype is linked to a specific pathogenic power or treatment sensitivity is a field of active research. However, there are still conflicting results. In addition, emerging strains might take benefit of the possibility of recombination to acquire resistance to the only available antiviral class of drugs or to escape from vaccine. Another important point relies on the fact that 78 % of chronic carriers live in Asia. This high percentage might by itself contribute to a high number of severe cases in this part of the world where genotype B and C are predominant. In the countries where genotypes A and D coexist, it has been suggested that patients infected by genotype A might evolve more rapidly during chronic infection than those infected with genotype D. Furthermore, different studies seem to sustain that patients infected with genotype C would progress to cirrhosis and liver cancer earlier than those infected by genotype B at the age of 30; however, at the age of 45 years, the same proportion of patients had been evolving to cirrhosis and liver cancer whatever the genotype was [15]. Other studies in the Amazonian basin indicated that the F genotype was frequently associated to severe acute hepatitis. In such cases, most of hospitalized patients were co- or superinfected by the hepatitis delta virus genotype 3 (HDV-3).
In contrast, several authors have found no link between HBV genotypes and the severity of hepatitis. For example, a study conducted in Uzbekistan didn’t demonstrate any difference of the severity of the liver histology between infections by HBV genotype D and A. Another recent study considering all case of liver damage (asymptomatic HBsAg carriers, acute or chronic hepatitis, cirrhosis, and hepatocellular carcinoma) also found no concrete link between genotype A or D and the severity of the hepatitis or response to treatment [38]. In summary, if there is not yet enough clear data to attribute to a specific genotype a clear predictive severity value, there is increasing evidence that in Asia, genotype C might be more frequently associated to HCC than genotype B. It is also important to evaluate these genotypes in function of the therapeutic response as this could represent predictive criteria. In a trial of PEG-IFN, patients infected with genotypes A and B had a higher rate of HBeAg loss (about 45 %) as compared to patients infected with genotype C or D (about 26 %).
4 Role of the HBx Protein
HBV encodes a small regulatory protein known as the HBx protein that stimulates HBV viral transcription and replication. HBx is a small polypeptide of 154 amino acids (16.5 kDa) encoded by the X gene which is highly conserved among mammalian hepadnaviruses. Designed as a probable “viral oncoprotein,” HBx favors HBV virus replication by exerting pleiotropic activities, such as destabilization of orderly cellular functions of cell cycle regulation, deregulation of different signaling pathways and DNA repair. Accumulation of such dysfunctions and alterations in cells may lead to viral persistence and hepatocarcinogenesis. In fact, during natural HBV infection, HBx is essential to initiate and maintain HBV [16]. In spite of HBx low expression levels both in acute and chronic infections, HBx induces humoral and cellular immune responses [17].
4.1 Epigenetic Modifications of Cellular Genes Due to HBx Protein
Many etiological forms of HCC focus on alterations that may take place at an epigenetic level [18]. Changes such as DNA methylation, histone modifications, and RNA-mediated gene silencing are largely responsible for HCC which lead to inactivation of tumor suppressor genes or chromosomal instability [19].
4.1.1 Modification of Gene Methylation
HBx acts as transactivator to activate its own promoter. Although considered as a weak transactivator, HBx is capable of activating a large number of cellular promoters. HBx is known to significantly increase the expression of genes by hypomethylating gene promoters of tumor-promoting genes. Such genes are retinal dehydrogenase 1, plasma retinol-binding protein precursor, and cellular retinol-binding protein I [20].
As demonstrated by chromatin immunoprecipitation analysis, HBx cannot bind directly to promoters of target genes.
However, the ability of HBx to transactivate a large number of cellular promoters can be explained by the direct interaction of HBx with DNMT3A (DNA methyltransferase 3A). In 2009, it was put into evidence that the expression of CDH6 and IGFBP3 was upregulated by the removal of DNMT3A from these promoters via the HBx-DNMT3A interaction [21]. Moreover, in the presence of HBx, the expression of DNMT1 and DNMT3A was also revealed to be upregulated. DNMT1 and DNMT3A are both responsible for the hypermethylation of gene promoters, implied in tumor suppression followed by gene silencing. The p16INK4A gene that regulates the cell cycle negatively undergoes such aberrant hypermethylation of CpG islands in the presence of HBx [22].
Contrary to the potential of HBx to transactivate genes, HBx is also capable of silencing the transcription of interleukin-4 receptor and metallothionein-1 F by recruiting DNMT3A to their regulatory promoters. This recruitment was shown to be responsible for de novo DNA methylation of these promoters that results in gene silencing [21].
These subtle mechanisms could portray a key role in HBx-induced HCC via cellular epigenetic modulations.
4.1.2 Histone Remodeling in the Context of HBx Protein
Aberrant histone acetylation can disturb cellular gene expression profiles. Such disturbances caused by abnormal histone acetylations via histone acetyl transferases or histone deacetylases are usually correlated with HCC. HBx is largely involved in HBV-induced HCC pathogenesis by promoting histone acetylation of tumor-associated genes. HBx was shown to interact directly with CBP/p300 in vitro and in vivo, thus synergistically enhancing C-AMP response element-binding protein (CREB) activity. It was also shown that HBx physically invades the CREB-binding domain of CREB responsive promoters of genes such as interleukin-8 (IL-8) and proliferating cell nuclear antigen (PCNA). Hence, an increased recruitment of CBP/p300 to the promoters of these genes was observed in the presence of HBx. The deregulation of these components can explain the neoplastic transformation of liver cells. For instance, IL-8 is a leukocyte chemotactic molecule that is upregulated in many human cancers, notably in liver cancer by its maintenance of an inflammatory state during the HBV infection. PCNA is involved in DNA replication and DNA repair, implying the potential contribution to cellular transformation when unbalanced [23].
Gene expression can also be modulated through induction of histone deacetylation. From recent findings, it was shown that HBx binds with histone deacetylase 1, thus involving itself in the repression of insulin growth-like factor binding protein 3 (IGFBP-3) [24]. The histone deacetylation of E-cadherin gene (CDH1) followed by hypermethylation by HBx silences the expression of CDH1. The mechanism involved in the loss of CDH1 is associated with various tumors and is present in numerous cancer cell lines. With reference to HBV, HBx was shown to form a complex together with mSin3A; thus, HBx downregulates E-cadherin expression by recruiting the mSin3A/histone deacetylase complex. Moreover, it was shown that HBx downregulated the expression of mir-373 known as a positive regulator of E-cadherin expression [25].
Aberrant histone methylations are also important modifications that occur in HBV-induced HCC pathogenesis. HBx upregulates the expression of SET and MYND domain containing 3 (SMYD3), which is a histone H3-H4-specific methyltransferase that is involved in the transcriptional activation of genes in relation to different cancers and, notably, HCC. The upregulation of SMYD3 during the HBV infection was shown to switch on the transactivation of the C-MYC oncogene. The latter is a famous oncogene, whose overexpression leads to the activation of various other genes promoting cellular proliferation and/or halting cellular differentiation. The HBx-related upregulation of C-MYC oncogene expression may occupy an important role in HCC development [26].
4.1.3 HBx and MicroRNA (miRNA)
HBx has also been shown to induce alterations on other epigenetic regulators such as miRNA. Such alterations of host miRNA profiles could be held responsible for the development of HCC. HBx was shown to upregulate miRNA 602 which inhibits the expression of the tumor suppressive gene RASSF1A [27]. Likewise, HBx was shown to increase strikingly the levels of miRNA-143 in HCC patients. This overexpression is mediated by the nuclear factor kappa B (NF-KappaB) repressing FNDC3B expression. The upregulation of miRNA-143 is in favor of the development of invasive tumors with metastatic properties [28].
Besides overexpressing certain miRNAs, HBx is involved in the downregulation of certain miRNAs. The let-7 family of miRNA which is generally highly expressed was shown to be downregulated in the presence of HBx. The downregulation of the let-7 family of miRNA by HBx was followed by a decreased expression of signal transducer and activator of transcription 3 (STAT3). The downregulated expression of STAT3 triggered cellular proliferation giving an additional indication of HBx deregulating cellular proliferation [29].
HBx was also shown to downregulate microRNA-101. The same downregulation was also put into evidence in HBV-related HCC tissues. DNA methyltransferase 3A which is a direct target of miR-101 is upregulated in the absence of miR-101; this upregulation in HBx-expressing cells increased the DNA methylation of tumor suppressor genes. Similarly, HBx was shown to downregulate the expression of microRNA-152 (miR152) in HBV-related HCC tissues. This downregulation leads to a heightened expression of DNA methyltransferase 1 (DNMT1) followed by a DNA hypermethylation of tumor suppressor genes such as glutathione S-transferase pi 1 (GSTP1) and E-cadherin (CDH1) [30].
Hence, HBx may regulate the cell epigenetic regulation through miRNA-related mechanisms. More studies are needed to explain the exact role of HBx together with different miRNAs. Whether HBx acts via a complex and consequent series of different cofactor recruitments to modify gene expression is a highly probable phenomenon.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


