Fig. 1
Simplified life cycle of HIV. The viral envelope, gp120-gp41, attaches to CD4 and a chemokine coreceptor, usually CCR5, and then fuses with the cell membrane. The HIV RNA genome is released in the cytoplasm, where it is reverse transcribed to a DNA copy that is transported into the nucleus where it is integrated into the host genome as the HIV provirus. Expression of HIV genes yields copies of the viral proteins and RNA genome that are exported to the cytoplasm, assembled into virions at the cell membrane, and budded into daughter virions
Some of HIV’s life cycle stages have been effectively targeted by the 2 dozen antiretroviral therapy (ART) drugs that are currently approved for the treatment of HIV, leading to partial recovery and markedly improved survival. However, because cells with integrated HIV proviral genome cannot yet be eradicated, treatment must be continuous and potentially lifelong.
It has been established that HIV is not able to induce malignant transformation, but promotes the effects of oncogenic viruses. This is achieved through compromising immune surveillance against infectious agents as well as against the cells displaying malignant characteristics. Another contribution is brought by the chronic hyperactivity of the immune system seen in the earlier stages of HIV infection. The excessive proliferation of the immune cells is coupled with an increased replication of the oncogenic viruses within those cells. Ineffective immune response could be the reason why other cancers, not related to infections, are encountered more often in people living with HIV [1].
3 Overview of Cancers in People with HIV
In most populations with HIV, cancer risk is dominated by Kaposi sarcoma (KS) and non-Hodgkin lymphoma (NHL). Effective ART reduces the risk for KS and NHL and increases longevity, such that other malignancies emerge. The risk factors and mechanisms for the most important malignancies among adults with HIV are summarized below. Malignancies in children are covered in Chap. 53.
4 Kaposi Sarcoma (KS)
KS is a malignancy of poorly differentiated, reprogrammed lymphatic endothelial cells particularly in the dermis [2]. KS typically appears as one or more red or violaceous skin lesions. Progression can include skin lesions that are widely disseminated and occasionally massive, lymphatic lesions that can cause lymphedema, mucosal lesions particularly on the hard palate and conjunctivae, gastrointestinal lesions that may bleed, and hepatic and lung lesions that result in death.
KSHV infection is necessary, but not sufficient, for the development of KS. KSHV primarily infects B lymphocytes and also T lymphocytes, macrophages, and the endothelial cells that may emerge as KS. Like EBV, the other human gammaherpesvirus, KSHV is primarily transmitted by saliva, in which infected individuals have a high concentration of virions. Susceptible individuals have readily accessible B lymphocytes in crypts of the tonsils and Waldeyer’s ring. In the latent phase of its life cycle, KSHV DNA persists as a circular episome in the nucleus, where it is copied and transmitted to daughter cells during mitosis. When triggered into its lytic phase, the circular episome splits open and dozens of viral genes are transcribed resulting in multiple copies of the KSHV genome and proteins that are assembled into virions that kill the cell when they are released to propagate the infection. A simplified life cycle of the gammaherpesviruses, KSHV and EBV, is presented in Fig. 2.
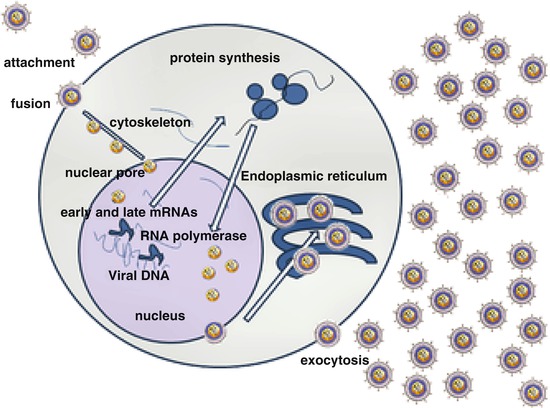
Fig. 2
Simplified life cycle of the gammaherpesviruses, KSHV and EBV. Virions are transmitted in saliva and primarily infect B lymphocytes in the tonsil. Following transit to germinal centers, viral DNA resides in the nucleus as an episome. During latency, few viral genes are expressed, but the viral genome is copied into daughter cells during mitosis. During the lytic phase, as illustrated, a cascade of viral genes is expressed resulting in release of daughter virions that kill the cell
A number of viral genes were identified, which may be responsible for the malignant transformation of the lymphatic endothelium induced by KSHV. Some of these have no correspondent in the target cells: K1, kaposin, and viral G protein-coupled receptor. Other viral genes generate proteins similar to those of human origin, thus deregulating the processes of cell multiplication and apoptosis. These viral genes include interleukins IL-6 and IL-10, CC-class chemokines, and the FLICE-inhibitory protein that blocks apoptosis. Two additional genes, latency-associated nuclear antigen (LANA) and K15, are involved in maintaining the persistence of the virus through the process of cell mitosis [3].
The prevalence of KSHV differs greatly between populations, being common in much of sub-Saharan Africa and in homosexual men in developed countries, rare in most of Asia, and intermediate in the Mediterranean region. The incidence of HIV-associated KS reflects these differences in KSHV prevalence.
Importantly, the risk for KS and its progression also are directly related to the severity of the immune deficiency, typically measured as the CD4 cell count in peripheral blood. Effective ART greatly reduces, but does not eliminate, the risk for developing KS. Effective ART also may be curative for early stage HIV-associated KS and improves the likelihood of response to chemotherapy for more advanced stages. Anti-herpesvirus drugs have not been proven effective in preventing or treating KS.
5 Non-Hodgkin Lymphoma (NHL)
High-grade, aggressive NHL, especially diffuse large B-cell lymphoma (DLBCL), primary central nervous system lymphoma (PCNSL), and Burkitt/Burkitt-like lymphoma are considered AIDS-defining among people with HIV. After KS, they have been the most common malignancies among people with AIDS.
Differences among these subtypes of AIDS-related NHL are noteworthy. PCNSL predominantly occurs with extreme immune deficiency (e.g., CD4 lymphocyte count <50/μL), is associated with very high mortality, and nearly always contains EBV in the malignant cells. Burkitt lymphoma can occur across the immune deficiency spectrum, even with CD4 lymphocyte count >500/μL and in people on effective ART [4]. Thirty to forty percent of AIDS-associated Burkitt lymphomas contain EBV. DLBCL is predominantly seen in people with a CD4 lymphocyte count <200/μL and may contain EBV in 30–80 % of cases [5].
Another, much less encountered NHL is the primary effusion lymphoma, characterized by a body serous cavity (pleura mostly) effusion as its main clinical sign. This malignancy appears in advanced AIDS and always contains KSHV, but EBV is also present in 80 % of cases. The mechanism of cooperation of the two viruses in the oncogenesis of the primary effusion lymphoma is not yet established [5].
The role of EBV in the pathogenesis of AIDS-related NHL is highlighted by a number of significant findings. EBV is one of the most widely spread viruses in humans, as it infects around 95 % of adults worldwide. As illustrated in Fig. 2, the lytic portion of the EBV life cycle can be triggered, leading to generation of daughter virions and infection of other susceptible cells or people. In contrast to the lytic cycle, for most of the time, EBV is present in latent form, mainly as a circular episome of DNA in the nucleus of B lymphocytes that replicates during cellular mitosis. Only a few viral genes are expressed during the latency phase, producing proteins that facilitate replication during mitosis but that also have the potential to support malignant transformation. EBV DNA also can be integrated into the host genome of malignant cells. The genetic lesions identified in EBV-infected malignant cells include c-myc gene rearrangement, bcl-6 gene rearrangement, ras gene mutations, and p53 mutations/deletions [3]. Three distinct patterns of viral gene expression have been identified during latency, each of them associated with different types of B-cell malignancies.
Perhaps the best understood oncogenetic process is the one which engenders the Burkitt lymphoma. The same genetic anomaly is present in all Burkitt tumors, namely, a translocation that brings the c-myc oncogene from chromosome 8 to either chromosome 2, 14, or 22, next to genes participating in the synthesis of immunoglobulins (Ig). In its new locus, c-myc is copied without restrictions whenever the synthesis of Ig is triggered. It important to note that this event is random and, in fact, that the cells containing this defect would undergo apoptosis sooner than the normal ones. However, EBV-infected mutated cells live longer, and those containing a particular mutation in the EBV genome, even much longer. Since not all Burkitt lymphomas contain EBV genomes, other mechanisms to ensure the protection from apoptosis of the mutated B lymphocytes may exist. This sequence of events is thought to constitute the first step in the development of Burkitt lymphoma [5]. As discussed above (see HIV epidemiology and pathogenesis), the role played by HIV in this process is to indirectly increase the chance of a spontaneous c-myc gene move to a new locus. This is achieved by inducing a massive B-cell proliferation in the early stages of the infection.
6 Hodgkin Lymphoma
HIV-associated Hodgkin lymphoma behaves like an AIDS-related condition, although it does not meet surveillance criteria for AIDS. Differences between HIV-associated and general population Hodgkin lymphoma are noteworthy. HIV-associated Hodgkin lymphoma is usually widely disseminated at diagnosis, and the tumors almost always contain EBV. The histologic subtype is predominantly mixed cellularity or lymphocyte depleted, whereas nodular sclerosis subtype is distinctly uncommon with HIV. The highest risk for HIV-associated Hodgkin lymphoma, especially mixed cellularity, is with moderately severe immune deficiency, with CD4 lymphocyte count 100–200/μL [6, 7]. It is possible that some cases of severe lymphocyte-depleted Hodgkin lymphoma are mistakenly diagnosed as NHL.
While the mutations in the B lymphocyte leading ultimately to the development of Hodgkin lymphoma are different from those in Burkitt lymphoma, the roles played by HIV and EBV are thought to be similar in both malignancies [5].
7 Squamous Cervical and Anal Cancers
Persistent infection with oncogenic types of HPV is the underlying cause of these malignancies, but accumulating evidence points to an important contributory role for HIV. In the general population, HPV is a very frequent sexually transmitted infection. The prevalence of HPV in the US female population aged 14–59 years was 42.5 % for the period 2003–2006 [8]. A review of the literature on HPV prevalence among men in Europe found a lower figure, 12.4 %, but in high-risk men, it was around 2.5 times higher, at 30.9 % [9]. Prevalence rates as high as 92 % have been found in HIV-positive men having sex with men (MSM) in San Francisco [10].
HPV, a DNA virus, has over 120 genotypes identified so far, but most of these are not oncogenic and HPV genotypes 16 and 18 account for around 70 % of cervical cancers and a higher fraction of anal cancers. Oncogenesis requires insertion of the HPV genome into the host cell’s DNA. A number of viral oncogenes have been identified; their mechanism of action is not entirely known. More specifically, three oncoproteins of viral origin, E5, E6, and E7, are responsible for interfering with cell multiplication controls. One such interference may be the inhibition of p53 protein with resulting suppression of apoptosis in the cells involved [11].
People with HIV are more likely to have prevalent anogenital HPV infection and, at least those with severe immune deficiency (CD4 lymphocyte count <200/μL), are less likely to clear an anogenital HPV infection compared to people without HPV [12, 13]. In the presence of HIV infection, the longer persistence of HPV increases the risk for squamous intraepithelial lesions of the uterine cervix. Incident HPV-related abnormal cervical cytology is less likely to occur and, if it does occur, more likely to resolve with the use of ART [14]. Compared to the general population, the incidence of invasive cervical cancer was nearly sixfold higher in women with AIDS [15], a portion of this excess related to the higher prevalence of HPV in women with HIV/AIDS. Likewise, compared to HIV-infected women with a CD4 count above 500 cells/μL, invasive cervical cancer was two- to threefold higher with a CD4 count of 200–499 cells/μL and 7.7-fold higher with a CD4 count <200 cells/μL [16].
Anal cancer is likewise an important morbidity for people with HIV, especially MSM. The incidence of anal cancer has been increasing in the general population, doubling over the past 30 years in the USA in both men and women [17]. On top of this increase, anal cancer incidence has been approximately 30-fold higher for people with AIDS [15]. The roles played by HPV and HIV in the pathogenesis of anal cancer are thought to be similar with their involvement in the cervical carcinoma.
The primary prevention of cervical carcinoma, and presumably of anal cancer too, is at present possible by means of HPV vaccination. Bivalent vaccines (against HPV 16 and 18) and quadrivalent ones (against HPV 16 and 18 and also HPV 6 and 11, known to produce genital warts) have been shown to prevent almost entirely the squamous intraepithelial lesions that precede invasive carcinoma [18, 19]. The population target of these vaccines, ideally, would be prepubertal and adolescent girls and boys. The immunization is safe and immunogenic in HIV-infected people too, as long as they still have the capability of producing a durable immune response [20].
The vast majority of the world’s population is not vaccinated, though. The Papanicolaou (pap) smear, combined or not with detection of HPV DNA in samples of cervical cells, remains useful in revealing the intraepithelial squamous lesions, precursors of invasive cervical cancers which are present for years before invasion begins. Such precursors, once localized by colposcopy, can be resected or destroyed. Where pap smears or colposcopy cannot be performed due to lack of resources, a simple visual inspection of the cervix after application of 5 % acetic acid can reveal areas of intraepithelial neoplasia and guide their treatment as above. Due to the faster progression of intraepithelial neoplasia to invasion in women with HIV, pap smears should be repeated yearly, as opposed to 3 years for the general population. With limited resources, this frequency may not be sustainable, but even offering three smears over a lifetime has the potential to halve the number of invasive cancers in the general population. Pap smears have not been evaluated for anal cancers. Here, as the disease is rare in the general population, only individuals at risk would require screening, which, at this time is possible only by anoscopy.
Lastly, it is noteworthy that the incidence of invasive cervical cancer with HIV, although it remains much higher than in the general population, decreased between 1996 and 2010 in the USA [21]. Whether this improvement reflects use of ART or improved cervical screening of women with HIV is unknown. No equivalent decline in anal cancer was observed [21].
8 Hepatocellular Carcinoma (HCC)
The current status of HCC with HIV was well reviewed by Sulkowski [22]. Irrespective of HIV, most cases of HCC are the result of chronic infection with either hepatitis B virus (HBV) or hepatitis C virus (HCV). HBV infection affects about one-third of the planet’s population, but only 6 % ultimately become chronic carriers, which nevertheless adds up to 350 million people. HCV infection is less widespread, involving about 3 % of the world’s population, but the ratio of chronic infection is higher than for HBV, estimated at between 55 and 85 % of those contaminated. Chronic hepatitis C is gaining more importance in the etiology of HCC as hepatitis B is preventable by vaccination. Moreover, HCV is usually acquired, at least in the USA and Europe, in the same way as HIV, by transfusion of blood from unscreened donors or by needle-sharing among drug users. In these populations, about 16 % of HIV-positive people also carry HCV [23].
The mechanisms by which HBV and HCV produce cancer in the liver are not elucidated, but they may be different. HBV is a DNA virus and integrates its genome in that of the hepatocyte. In contrast, HCV is an RNA virus, but not a retrovirus and thus without a nuclear intermediate. Factors identified to contribute to oncogenesis by HCV are deregulation of cell cycle proteins, interference with p53 activity, and chronic inflammation.
HCV infection is more likely to persist indefinitely, with high levels of replication and viremia, in people who have also been infected with HIV. Such chronic HCV infection increases the risk for HCC. The contribution of HIV consists, on the one hand, in degrading the innate immunity of the host, which normally would play an important role in clearing HCV infection, by means of increased production of interferon-gamma. On the other hand, depletion of CD4+ T-cells by HIV reduces the antibody response to HCV and thus additionally facilitates the persistence of the latter. Further, a variant in the interferon λ-4 gene, while facilitating clearance of HCV infection, may paradoxically shorten the survival of HIV-infected individuals who receive ART [24–26]. HCC morbidity and especially mortality with dual HCV-HIV infection is very high [27, 28]. Newly developed direct-acting antiviral agents offer the hope that HCV can be effectively treated prior to progression to HCC, even with HIV coinfection [29–31].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


