Constitutional and Acquired Autosomal Aneuploidy
Departments of Pathology and Human & Molecular Genetics, Virginia Commonwealth University, Richmond, VA 23298, USA
Key Words
• Aneuploidy • Mosaicism • Acquired chromosomal aneuploidy • Epigenetics • Trisomy • Age-related aneuploidy • Chromosomal instability
The consequences of chromosomal abnormalities have been noted “from the earliest stages of life well through the reproductive years, and even beyond.”1(p7) These chromosomal imbalances can result from numerical or structural anomalies. The numerical chromosomal abnormalities are often referred to as aneuploid conditions. The Greek translation of the word aneuploidy is “not a good set.” Thus, aneuploidy refers to a chromosomal number that is not an exact multiple of the haploid complement (ie, in humans 45 or 47 chromosomes per cell). Aneuploidy can arise in gametes/embryonic cells (constitutional) or in differentiated somatic cells (acquired) and can involve autosomes (chromosomes 1–22), as well as gonosomes (chromosomes X or Y). This article focuses on the occurrence of constitutional and acquired autosomal aneuploidy in humans.
Constitutional Autosomal Aneuploidy
Frequency of Aneuploidy
The earliest cytogenetic studies, which were completed in liveborns, showed aneuploidy involving only 3 of the 22 autosomes, with only trisomy (no monosomy) conditions being seen.2,3 Although cytogenetic methodology has advanced, the results of these pioneering investigations have remained relatively unchanged. The most frequently occurring autosomal aneuploidy condition noted in newborns is trisomy 21, which is seen in approximately 1 in 830 livebirths2,3 (Table 1). The other autosomal trisomies that are seen in newborns include trisomy 18 (observed in approximately 1 in 7500 livebirths), trisomy 13 (approximately 1 in 22,700 livebirths), and very rare cases of trisomy 22.2–4 Soon after the discovery of these autosomal aneuploidy conditions, cytogeneticists conjectured that the incidence of aneuploidy in humans was not limited to primarily three chromosomes (13, 18, and 21), but could occur for all autosomes, with the clinical consequences of aneuploidy for the other autosomes being incompatible with life. To test this hypothesis, investigations were initiated to determine if autosomal aneuploidy was present in spontaneous abortus tissue.5 Chromosomal analyses of products of conception (primarily first trimester) showed the presence of trisomies involving nearly all autosomes. Interestingly, trisomy for chromosome 16 accounted for approximately a third of all autosomal trisomic first-trimester abortuses5 (see Table 1). Other autosomes that were observed to have increased frequencies of trisomy in first-trimester abortus tissue included chromosomes 2, 7, 8, 18 and the acrocentric chromosomes (13, 14, 15, 21, and 22; see Table 1). Explanations for these observed interchromosomal differences included (but were not limited to): (1) a true difference in the propensity for a chromosome to undergo malsegregation; or (2) a selection bias that reflected the differential viability of imbalances involving different chromosomes, or both. To distinguish between these plausible explanations, aneuploidy data were collected directly from gametes.
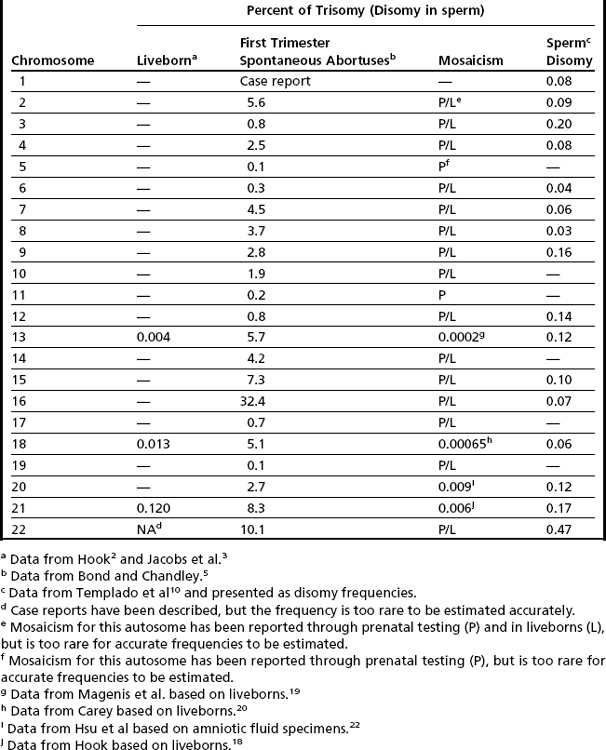
The earliest of the gametic aneuploidy studies directly analyzed metaphase chromosomes from oocytes or sperm (Fig. 1). More recently, researchers have completed these investigations using DNA-based methods (primarily fluorescence in situ hybridization [FISH], but also comparative genomic hybridization techniques).6–8 Collectively, the results of these gametic studies have shown that humans have a relatively high frequency of gametic aneuploidy compared to the frequencies seen in other species.9 Moreover, the frequency of sperm aneuploidy has been shown to differ between chromosomes (see Table 1), suggesting that at least a portion of the observed variation in autosome-specific frequencies of aneuploidy reflects true differences in autosomal malsegregation, rather than being limited solely to selective growth pressures.6,10 Gender differences have also been noted in gametic aneuploidy frequencies, with oocytes having higher levels of aneuploidy than sperm.6–8 However, technical challenges have thwarted investigators’ abilities to provide unbiased, chromosome-specific estimates of autosomal aneuploidy present in oocytes.11,12 One factor that is thought to contribute to the observed overall increased frequency of aneuploidy in oocytes compared to spermatocytes in humans is their means of ascertainment. The majority of oocytes available for study are ones found unsuitable for transfer from assisted reproductive procedures (such as in vitro fertilization), thus leading to biases associated with maternal age (see later) or with the presence of morphologic anomalies.8 However, studies of nonhuman mammals, which are not limited by these ascertainment biases, have also shown significant increases in the frequency of aneuploidy in oocytes compared to spermatocytes.13,14 Etiologic factors that have been suggested to contribute to the higher incidence of aneuploidy in oogenesis include (but are not limited to) (1) the disruption of the meiotic division in prophase I (with the division starting prenatally, but not commencing until ovulation, which may occur decades later); (2) differences in meiotic checkpoint permissiveness for errors in males compared to females; (3) differences in the frequency and chromosomal location of meiotic recombinational events (see discussion of etiology later); and (4) epigenetic differences in response to environmental/stressor exposures.
Mosaicism for Autosomal Aneuploidy
Mosaicism for aneuploidy is defined as the presence of two or more distinct cell lines that are derived from a single zygote but differ from one another because of nondisjunction (or mutation).15 The use of interphase FISH techniques has allowed for improvements in the ability to detect autosomal mosaicism because thousands of interphase nuclei from a variety of different tissues can be evaluated (not limited only to viable cells that can be successfully cultured in vitro to produce metaphase spreads).16,17 Mosaicism for nearly all of the autosomes has been identified in either liveborns (usually ascertained because of health/developmental concerns) or in fetuses through prenatal testing (asymptomatically or due to ultrasound aberrations; see Table 1).18–22 Given that most of the instances of trisomic mosaicism in liveborns are case reports, unbiased estimates of the frequency of mosaicism for all of the autosomes are not available. However, mosaicism for trisomy 21, which is thought to be one of the most frequent autosomal mosaicism conditions in liveborns, is seen in only 2% to 5% of all people with Down syndrome (or approximately 1 in 16,670–41,670 livebirths).18 A caution to be noted when estimating frequencies of mosaic conditions in liveborns is that there is likely to be a bias toward studying only individuals who have clinical consequences resulting from their mosaicism. Thus, the estimates based on case reports/clinical studies are likely to be underestimates of the true frequency of constitutional autosomal mosaicism in humans.
The overall frequencies of true autosomal mosaicism (not pseudomosaicism) observed through prenatal testing is approximately 0.2% after amniocentesis and 1% to 2% after chorionic villi sampling (CVS), with the latter figure including both mosaic complements and nonmosaic complements from direct preparations that differed from those of the fetus (indicating confined placental mosaicism).22,23 Chromosome-specific differences in the frequency of prenatally diagnosed autosomal mosaicism have been identified, with trisomies involving chromosomes 13, 18, 20, and 21 being seen most frequently in amniocytes.24,25
One means for gaining insight regarding the frequency of mosaicism in humans has been to study early embryos, which were obtained after assisted reproductive procedures. These embryos have shown an unexpectedly high frequency of mosaicism, with the majority (50%–91%) of early (day 4) embryos having mosaicism involving single or multiple (sometimes referred to as chaotic embryos) chromosomes.26,27 Chromosomal mosaicism has also been observed in fetal (30%–35% of cells) and adult (10%) brains cells28,29; liver cells (approximately 3% in adults)30; and other tissues,31 including germline mosaicism, which, if present, could have reproductive consequences.32 These observations have led investigators to postulate that mosaicism may occur as a “normal” or global biological process in early human development, with tissue-specific selection leading to loss of most aneuploid cells in the early stages of development, but with some tissues “normally” retaining mosaicism.31 Iourov and colleagues31 further speculate that mosaicism (often undetected) may contribute to the development of several conditions (including autism, schizophrenia, autoimmune diseases, and Alzheimer disease), with a latent, age-related influence of mosaicism being conjectured for a portion of these conditions.
Phenotypic Findings Associated with Constitutional Mosaicism
As expected, because individuals with trisomy for chromosomes 13, 18, or 21 in every cell can survive to term, fetuses having mosaicism for these conditions can also survive. The phenotype of these individuals ranges from near normal to the demonstration of a subset (or in some cases all) of the phenotypic traits that have been well characterized in people having Patau, Edward, or Down syndrome.16,33,34 Individuals with mosaicism for trisomy 21 have phenotypic variation that appears to be influenced by the proportion of cells having a trisomic (compared to a euploid) complement, as well as tissue-specific effects.16 However, positive karyotype–phenotype correlations have not consistently been seen in people having mosaicism involving other trisomic conditions.33,34 As a result, the percentage of trisomic cells present in an individual having mosaicism cannot be clearly used as a prognostic indicator.
Interestingly, many individuals having mosaicism (regardless of the chromosome involved) have skin pigmentary abnormalities, which may not be present until weeks or years after birth; intrauterine growth compromise; body/facial/limb asymmetry; and developmental delay. As a result of this phenotypic overlap, the term “general chromosomal mosaic syndrome” has been coined.35 However, for a subset of these conditions, specific syndromic patterns have emerged (summarized in Table 24,16,21,23–25,33–75), thereby facilitating the recognition of the chromosomal basis for these individual’s health/developmental problems. The provision of genetic counseling after a prenatal diagnosis of autosomal mosaicism can be especially challenging. Genetic counseling concerns for these patients include (1) variation in phenotypic consequences ranging from the extremes of a normal outcome to a severely abnormal phenotype; (2) identification/confirmation/quantitation of the trisomic cell line, with fibroblasts (and placenta) tending to show the presence (or higher frequencies) of the trisomic line more often than blood; (3) an assessment of the possible presence of uniparental disomy in the euploid line, due to a trisomy (or monosomy) rescue mechanism (particularly in cases where mosaicism was detected through CVS and in cases where the trisomic [monosomic] chromosome is one known to have imprinted regions); (4) recognition that clinical outcome can vary based on the mode of ascertainment (eg, was the mosaicism detected through CVS [if so, is it confined placental mosaicism?] vs amniocentesis, or after birth [due to phenotypic findings]); and (5) the technical criteria that were used to define the mosaicism (true mosaicism vs pseudomosaicism).22 For mosaic cases ascertained after prenatal testing, one may wish to recommend a fetal scan to potentially detect phenotypic abnormalities and to advise that follow-up confirmation chromosomal testing be completed (if CVS, then confirm through amniocentesis; if amniocentesis, then confirm through percutaneous umbilical blood sampling [for cases in which the trisomic line can be seen in blood] or at termination/birth).37
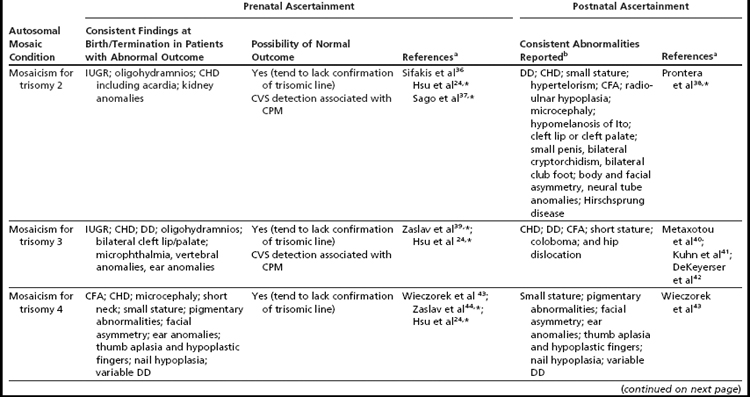

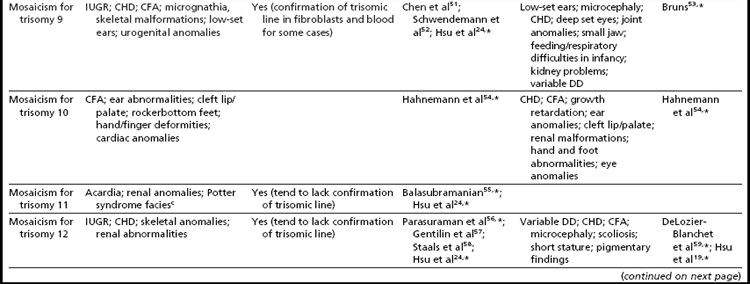
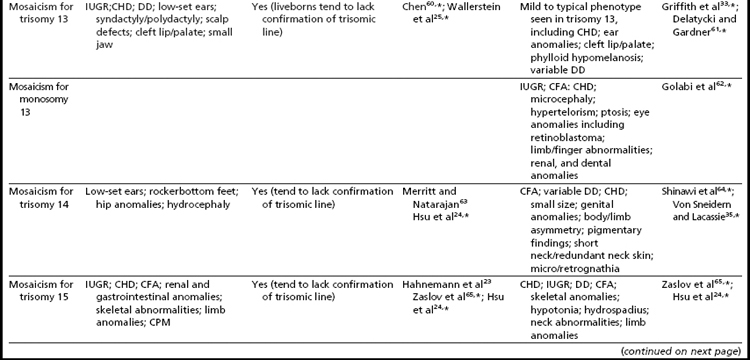

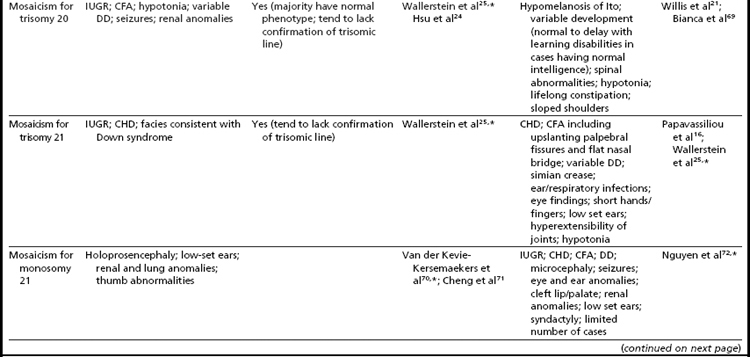

In addition to mosaicism involving autosomal trisomy, patients having mosaicism for autosomal monosomy have also been described, including mosaic monosomies for chromosomes 13,62 21,70,72 and 224 (see Table 2). However, these conditions are too rare to allow for the establishment of a distinct syndromic phenotype. On a cautionary note, when feasible, the presence of an apparent autosomal monosomic cell line should be confirmed using molecular cytogenetic approaches, as several patients reported to have monsomic cell lines on the basis of GTG-banding were subsequently determined to have cryptic structural abnormalities (not full monosomy).72
Etiology of Constitutional Autosomal Aneuploidy
Having well established that constitutional autosomal aneuploidy was a significant factor leading to morbidity and mortality in humans, investigators initiated studies to understand the underlying causes of aneuploidy and to understand the basis for the observed differences in aneuploidy frequencies between chromosomes. Despite years of study, the etiology of aneuploidy in humans remains largely enigmatic, as does the specific mechanism(s) whereby the observed imbalances (trisomy) result in phenotypic consequences involving multiple organs. Investigators completing epidemiologic studies have consistently shown that advanced maternal age correlates with an increased risk for having a child with trisomy 21 (as well as most other trisomic conditions). This association has been observed in women from all ethnic and geographic backgrounds.76 Knowledge of other factors (or the mechanistic influence of maternal age) contributing to the propensity for human chromosomes to malsegregate in meiosis has come from studies of the parental origin of the extra chromosome in trisomic probands.77 Investigations of nuclear families (mother, father, and trisomic child) have shown that the majority (93%–95%) of nondisjunctional events leading to a child having trisomy 21 occurred in oogenesis (primarily meiosis I).77 Similar studies of nuclear families in which a child has Edward syndrome or Patau syndrome have also shown that gametic malsegregation occurred more often in oogenesis than in spermatogenesis. However, the malsegregational event that occurred in the gametes that resulted in the birth of a child with trisomy 18 occurred more frequently in meiosis II (58.7% of cases) than in meiosis I.77 Studies of abortus tissue (and parental samples) have revealed variation in the parental and meiotic origin of the malsegregational events for the different autosomes.77
Attributes that have been conjectured to influence a chromosome’s propensity to malsegregate include the presence of chromosomal heteromorphisms.78,79 However, because the association of these heteromorphisms with aneuploidy has not been consistently observed, their influence on chromosomal segregation, if any, seems to be either minimal or to act via a mechanism that is not straightforward (eg, increased amounts of heterochromatin may have epigenetic effects that, in turn, influence aneuploidy).79,80
Based on information gained from studies of the DNA markers evaluated in parental origin studies, perturbations in recombination have been recognized to occur in gametes that resulted in aneuploidy offspring/conceptuses.80 For trisomy 21, reductions in the number of chiasmata, as well as changes in their location, have been observed, with the results of more recent studies suggesting that these patterns may be influenced by age, as well as environmental factors.14,81 One can speculate that a lack of recombination (achiasmate bivalents) could lead to distributive segregation of the chromosomes, which is a phenomenon that has been shown to arise in Drosophila, but has not been definitively observed in humans.82 Simplistically, the hypothesis of distributive pairing can be likened to a “singles’ bar” (Fig. 2). In distributive pairing, the unpaired chromosome(s) are thought to be more likely to pair with chromosomes having a similar size. By chance, the unpaired homologs could pair with one another, leading to potentially normal segregation at meiosis. Alternatively, the achiasmatic chromosomes could pair with a nonhomologous chromosome. The segregation of these nonhomologous chromosomes could result in either a euploid or aneuploidy gamete (see Fig. 2). Aberrant localization of chiasmata between homologs can also lead to malsegregation, with a location that is too proximal resulting in entanglement whereas one too distal can result in unorganized, random segregation.80
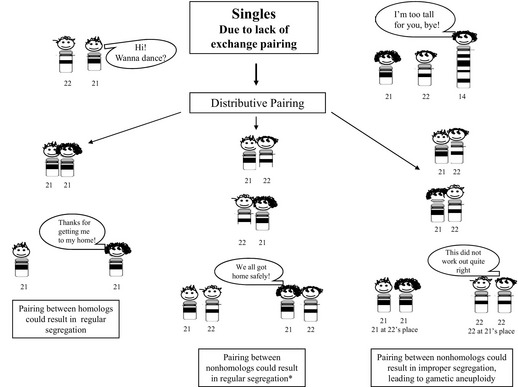
The “trigger(s)” inducing perturbations in recombination, or other mechanisms contributing to the genesis of gametic aneuploidy, have not been clearly recognized, but are likely to include both genetic and environmental influences (Fig. 3). Although studies of nonhuman eukaryotic species have allowed for the recognition of many genes that contribute to the proper segregation of chromosomes in meiosis, the a priori recognition of genetic changes leading to an increased risk for gametic aneuploidy in humans is still lacking. Genes that have been identified as likely candidates contributing to gametic aneuploidy include (but are not limited to) those involved in meiotic/spindle checkpoints, centrosome formation/duplication, chromatid cohesion, and chromatin organization.83–89 A “two-hit” model of aneuploidy has been proposed to integrate the maternal age influence with the effects from genetic and other potential etiologic factors that have been associated with aneuploidy in humans.90,91 This model postulates that the “first hit” is the presence of a primary oocyte(s) having a susceptible meiotic configuration (such as perturbations in recombination). If these susceptible oocytes experience a “second hit,” which could occur years later due to compromises in the functioning of key genes (noted previously), they could result in an aneuploid conceptus. Alternatively, the “second hit” could be related to environmental exposure(s).
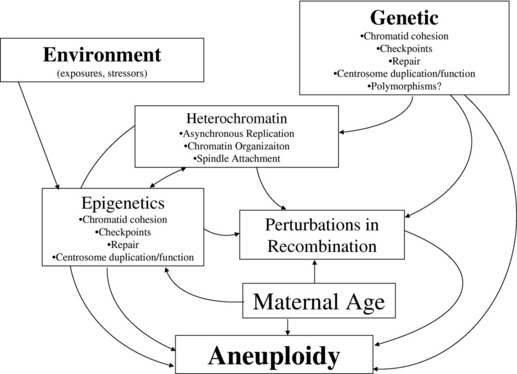
The designation of specific environmental exposures as aneugens in humans has been problematic, with both supportive and nonsupportive data reported for a variety of toxins/lifestyle exposures.92 In studies performed in mice, Hunt and colleagues93 clearly showed an increase in aneuploidy frequencies in response to bisphenol A (BPA) exposure. BPA, which is a xenoestrogen that activates estrogen receptors, has been conjectured to elicit its aneugenic effect through perturbations in the function of the tubulin gene, thereby disrupting spindle organization.94 BPA has also been shown to induce epigenetic changes in developing mammals that differ between species and vary with dosage.95 Excitingly, the biological response of mice to BPA can be modulated with diet, suggesting that the impact of this chemical (and possibly the effects of other aneugens) may have biological plasticity/reversibility.95,96
The field of epigenetics, which investigates heritable alterations in gene expression or phenotype that are caused by mechanisms other than changes in the underlying DNA sequence (eg, methylation changes, histone alterations, microRNA expression), is an exciting area of study that may allow for improvements in the understanding of etiologic factors contributing to aneuploidy.97 One can conjecture that epigenetic changes may explain, at least in part, the long recognized influence of advanced maternal age on chromosomal aneuploidy in humans. Epigenetic modifications could be acquired (over time) in gametes, leading to changes in the expression of genes that are involved in the resumption of meiosis I segregation or meiosis II segregation (including but not limited to genes involved in chromatid cohesion, meiotic/spindle checkpoints, centrosomes, spindle formation, DNA repair). These epigenetic modifications could result from a wide variety of “environmental” stressors (classic environmental exposures or social stresses) that may have occurred over many years, making their detection elusive (see Fig. 3). Their impact may also be modulated by diet or hormonal influences, thereby mimicking patterns consistent with those noted for complex diseases. Although the role of epigenetic changes on the initiation of chromosomal malsegregation is largely speculative, investigators have shown that epigenetic changes arise in response to trisomic imbalances and may also contribute to the variation in phenotypic findings present in people having chromosomal aneuploidy conditions.98
Studies of individuals having mosaicism may provide valuable insight about the causes of aneuploidy in humans. Although all cases of mosaicism require at least one postzygotic error, we (unpublished data) and other investigators99,100 have shown that in the majority of individuals having mosaicism for trisomy 21, two chromosome malsegregational events occurred (both a meiotic and a mitotic error), with a “trisomy rescue” scenario giving rise to the normal cell line. Thus, it is feasible that the chromosomes in these individuals (which malsegregated twice) have a biological basis for a predisposition to aneuploidy. From a genetic counseling perspective, this observation is important to recognize because the recurrence risks for couples having a child with mosaicism for trisomy 21 may be comparable to those of couples having a child with “complete” trisomy 21, rather than being negligible, as has been suggested in the early literature (probably as a result of the rarity of this condition, which prohibited the collection of robust empiric risk data).
Related posts:
 Cytogenetics
Cytogenetics
 Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
Structural Rearrangements: Detection and Elucidation of Mechanisms Using Cytogenomic Technologies
 Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
Evolving Picture of Microdeletion/Microduplication Syndromes in the Age of Microarray Analysis: Variable Expressivity and Genomic Complexity
 In Situ Hybridization
In Situ Hybridization
 Myeloma: Current Perspectives
Myeloma: Current Perspectives
 Syndromes
Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


