Mutation
Description
Missense
A change in one DNA base pair that results in the substitution of one amino acid
Nonsense
A change in one DNA base pair that results in the substituting of one amino acid that encodes a stop codon
Insertion
Changes in the number of DNA base pairs in a gene by inserting additional DNA base pairs
Deletion
Changes in the number of DNA base pairs in a gene by removing a piece of DNA
Duplication
A piece of DNA that is aberrantly copied one or more times
Frameshift
The addition or loss of DNA base pairs that changes the reading frame
Repeat expansion
Short DNA sequences that are repeated a number of times in a row
7.2 Proto-oncogenes
The discovery of proto-oncogenes is one of the most fundamentally important findings of this century. Oncogenes are activated (frequently mutated) alleles of normally functioning wild-type genes (proto-oncogenes) that function in cell cycle progression or cellular proliferation. Activated or mutated proto-oncogenes promote unregulated cell cycle progression and cell proliferation, leading to cancer development. Proteins encoded by normal cellular proto-oncogenes function in all subcellular compartments including the nucleus, cytoplasm, and cell surface, and exert their function in most intracellular processes by acting as protein kinases, growth factors, growth factor receptors, or membrane associated signal transducers. Mutations in proto-oncogenes alter the normal structure and/or expression pattern, and the resulting oncogene acts in a dominant manner. That is, a mutation in only a single allele is required for activation of the proto-oncogene and/or loss of regulation of the proto-oncogene product. In genetic terms, this is typically referred to as a gain of function mutation.
7.2.1 Viral Oncogenes
The discovery of proto-oncogenes is rooted in the study of mammalian viruses, and in particular, retroviruses. In the earlier years of the twentieth century, radiation, chemicals, and viruses were shown to induce cancer in experimental animals, and later, transform cells in culture. The study of so called tumor viruses advanced quickly compared to studies of radiation-induced and chemical-induced carcinogenesis, for several reasons. Although both chemical carcinogens and radiation are potent inducers of neoplasia, it was found that tumor viruses could more efficiently and reproducibly transform cells in culture and induce tumors in experimental animals. Tumor viruses caused tumors to develop in a matter of days to weeks allowing rapid analysis following infection. Moreover, both radiation and chemical carcinogens act randomly on the cellular genome. Examining the cellular genome to determine the carcinogenic effect of these agents at a level of individual genes or DNA segments was a daunting task. In contrast to the mammalian cellular genome, the small genome of the tumor viruses offered a less complex model for identification of specific sequences responsible for induction and progression of tumors, and a more efficient system for elucidation of molecular mechanisms governing neoplastic transformation.
7.2.1.1 DNA Viruses
Some of DNA viruses and one class of RNA viruses (the retroviruses) have been shown to have oncogenic potential. The Shope papilloma virus was one of the first DNA tumor viruses to be described [3]. It causes benign papillomas that can progress to malignant carcinomas in cottontail rabbits. Papillomaviruses, along with other classes of DNA viruses such as the hepatitis B viruses, have the ability to transform cells in their natural host. Most other DNA viruses, including adenoviruses, simian virus 40 (SV40), and polyomaviruses lack transforming ability. In natural hosts, cells infected with these DNA viruses undergo cell death rather than transformation as a consequence of viral replication. However, these later viruses demonstrate their oncogenic potential in heterologous, nonpermissive species in which viruses cannot replicate. Each class of DNA virus has led to remarkable discoveries in proto-oncogenesis [4].
7.2.1.2 RNA Retroviruses
The RNA retroviruses represent the class of tumor viruses that has contributed the most to our understanding of mammalian carcinogenesis. Retroviruses are the only currently known RNA viruses to have oncogenic potential. A feature common to these viruses is the ability to replicate in infected cells via a provirus intermediate. The proviral intermediate is generated through the action of a retroviral enzyme termed reverse transcriptase, which synthesizes a DNA copy of the retroviral RNA genome. RNA to DNA reverse transcription is obligatory for RNA retroviral replication in infected mammalian cells. The DNA transcript of the retroviral genome incorporates into the cellular genome where it replicates along with cellular DNA. The RNA polymerase enzyme of the host cells transcribes the provirus DNA, generating new RNA virions and retroviral mRNAs needed for synthesis of viral proteins. Importantly, unlike most of the DNA viruses, retroviruses are not cytotoxic or cytocidal to the host cells. This reflects the nature of the retroviral lifecycle, where new retroviral particles are released from the cell by budding rather than by cell lysis. Thus, RNA viruses can transform the same cells in which they replicate. The recombination event that occurs between the retroviral DNA (provirus) and host DNA as part of the replication cycle has significant implications for neoplastic transformation and tumor development [5].
The first oncogenic retrovirus discovered was the Rous sarcoma virus (RSV) . Peyton Rous inoculated chickens with a chicken sarcoma cellular extract and was able to demonstrate efficient transmission of an agent that propagated tumor growth [6]. Subsequent studies demonstrated that RSV had transforming properties in cultured cells. This was found to be in contrast to another well-studied retrovirus, the avian leukosis virus (ALV) . ALV maintained the ability to induce tumors following inoculation in chickens (albeit after months, and not days to weeks compared to RSV), but did not demonstrate the ability to transform cells in culture [4]. The differences in induction efficiency in animals (in vivo activity) and ability to transform cells in culture (in vitro activity) form the basis for dividing retroviruses into two groups: (1) the acutely transforming oncogenic retroviruses, and (2) the weakly oncogenic or non-transforming retroviruses.
7.2.2 Cellular and Retroviral Oncogene Discovery
The differences between the acutely and weakly or non-transforming retroviruses are extremely important and provided clues towards recognition of the first proto-oncogene. In comparing RSV and ALV genomes, RSV was shown to be 1.5 kb greater in size than ALV. This additional segment was correctly postulated to be responsible for the rapid transforming properties of RSV. In 1971, Peter Vogt isolated RSV mutants that had the weakly oncogenic properties of ALV [7]. These weakly oncogenic RSV mutants were approximately the same size as ALV, did not have the ability to transform cultured cells, and did not efficiently induce sarcomas in animals, but maintained retroviral replication capabilities. The missing 1.5 kb sequence in these mutant RSV genomes was subsequently demonstrated to be required not only for initiation but also for maintenance of neoplastic transformation. Because different RSV mutants were not complimentary and did not lead to neoplastic transformation in cell culture, it was concluded that a single gene could be responsible for both in vitro transformation and in vivo oncogenesis. The first retroviral oncogene was named v-src for its sarcoma inducing action. Since then over 30 viral oncogenes have been discovered in over 40 transforming viruses [4, 8, 9].
Similar to the discovery of the first oncogene, the discovery of the origin of retroviral oncogenes had monumental implications. The extra 1.5 kb of nucleic acid in RSV was not necessary for viral replication/growth. It was not clear where the apparently extraneous nucleic acid segment originated. The answer was obtained through the study of retroviral tumors of the very rare animal that developed tumors after being infected by a non-transforming retrovirus. These previously non-transforming retroviruses were found to have incorporated new genetic material in their RNA genome corresponding to a new oncogene which conferred capability for neoplastic transformation. The portion of the proviral genome corresponding to the newly recognized oncogene was used to probe for similar sequences in host cells. This analysis demonstrated that genes possessing the capability for neoplastic transformation were conserved among several different species. This observation suggested that host cell DNA could be incorporated into the genome of a retrovirus during recombination in the infected cell. Further study of the cellular homologues of retroviral oncogenes showed that they are normal cellular genes that encode proteins involved in various aspects of cellular homeostasis, including cell proliferation and differentiation. The normal cellular counterpart of the retroviral oncogenes is referred to as cellular proto–oncogenes. The current paradigm holds that viral oncogenes originate from cellular proto-oncogenes, and that these genes have been altered in a manner which confers the ability to induce cellular neoplastic transformation in infected cells [4, 8]. In like manner, cellular proto-oncogenes can be activated in various ways (point mutation, deletion, amplification, or rearrangement) that result in the synthesis of an oncogenic protein product [4, 8].
The discovery of the ability of genes to induce tumors in animals and humans linked the study of transforming retroviruses with the field of molecular biology of human cancers. However, it is clear that most human cancers are not caused by infection with transforming viruses. Shortly after it was established that specific virus associated genes could cause cellular transformation, alterations in cellular proto-oncogenes were found to be responsible for human tumors. The first instance linking the possibility of a human proto-oncogene with cancer, when retroviral involvement could be eliminated, was reported in 1981 by two groups, who showed that DNA extracted from a human bladder carcinoma cell line (EJ) could induce transformation in an immortalized but non-transformed mouse cell line NIH 3T3 [9, 10]. In 1982, the first human activated proto-oncogenes were isolated and identified from the EJ bladder carcinoma cell line and a human lung carcinoma. These genes were cellular homologs of the Harvey-ras and Kirsten-ras retroviral oncogenes, both of which had previously been shown to induce rat sarcomas [11]. The discovery of proto-oncogenes solidified the link between genes and cancer, and ushered in an era of genetic discovery focused on identification of genetic abnormalities that contribute to the development of human neoplasms.
7.2.3 Mechanisms of Activation of Cellular Proto-oncogenes
Cellular proto-oncogenes must become activated in order to express oncogenic potential leading to neoplastic transformation. Activation of cellular proto-oncogenes typically involves chromosomal translocation, amplification, or point mutation. The changes that result can be broadly categorized into (1) changes to the structure of a proto-oncogene which result in an abnormal gene product with aberrant function (examples include the bcr-abl translocation and c-ras point mutations, described below) and (2) changes to the regulation of gene expression resulting in aberrant expression or inappropriate production of the structurally normal growth-promoting protein (examples include translocations involving c-myc, amplification involving N-myc in neuroblastomas, and some point mutations in c-ras).
7.2.3.1 Proto-oncogene Activation Through Chromosomal Translocation
Translocation leading to structural alteration of bcr-abl . Evolving techniques in cytogenetics over the last century have led to increased resolution of individual chromosomes. Abnormalities in chromosomes were known to occur in neoplastic cells from at least 1914, when Boveri noted somatic alterations in the genetic material of sea urchin eggs fertilized by two sperm. The abnormal cells looked similar to tumors, and he hypothesized that cancer might result from cellular aberrations that produced abnormal mitotic figures [12]. However, it was not clear whether chromosomal abnormalities represented primary oncogenic events, or accumulated errors secondary to neoplastic transformation. Initially, the plethora of chromosomal abnormalities favored the latter scenario, as no consistent chromosomal abnormality was identified upon examination of many tumors and similar tumors from different individuals. That changed in 1960 when Nowell and Hungerford described the first reproducible tumor-specific chromosomal aberration in chronic myelogenous leukemia [13]. They observed the presence of a shortened chromosome 22, subsequently named the Philadelphia chromosome after the city in which it was discovered. It was found in cancer cells from over 90 % of patients with chronic myelogenous leukemia (CML). This observation suggested that (1) the abnormality may have imparted some form of growth advantage over other cells and may be causally related to the development of tumors, and (2) other neoplasms may also harbor their own specific chromosomal or genetic aberrations. Since the first recognition of common chromosomal abnormalities in specific human tumors, numerous translocations involving important genes have been characterized (Table 7.2).
Table 7.2
Chromosomal translocation breakpoints and genes
Type | Affected gene | Disease | Rearranging gene | ||
|---|---|---|---|---|---|
Non-fusions/hematopoietic Tumors | |||||
Basic-helix-loop-helix | |||||
t(8;14)(q24;q32) | c-myc (8q24) | BL, BL-ALL | IgH, IgL | ||
t(2;8)(p12;q24) | |||||
t(8;22)(q24;q11) | |||||
t(8;14)(q24;q11) | c-myc (8q24) | T-ALL | TCRα | ||
t(8;12)(q24;q22) | c-myc (8q24) | B-CLL/ALL | |||
BTG (12q22 | |||||
t(7;19)(q35;p13) | lyl1 (19p13) | T-ALL | TCRβ | ||
t(1;14)(p32;q11) | tal1/SCL | T-ALL | TCRα | ||
t(7;9)(q35;q34) | tal2 (9q34) | ||||
LIM proteins | |||||
t(11;14)(p15;q11) | RBTN1/Ttg1 (11p15) | T-ALL | TCRδ | ||
t(11;14)(p13;q11) | RBTN2/Ttg2 (11p13) | T-ALL | TCRδ/α/β | ||
t(7;11)(q35;p13) | |||||
Homeobox protein | |||||
t(10;14)(q24;q11) | hox11 (10q24) | T-ALL | TCRα/β | ||
t(7;10)(q35;q24) | |||||
Zinc-finger protein | |||||
t(3;14)(q27;q32) | Laz3/bcl6 (3q27) | NHL/DLCL | IgH | ||
t(3;4)(q27;p11) | Laz3/bcl6 (3q27) | NHL | |||
Others | |||||
t(11;14)(q13;q32) | bcl1 (PRAD-1) (11q13) | B-CLL and others | IgH, IgL | ||
t(14;18)(q32;21) | bcl2 (18q21) | FL | TCR-Cα | ||
inv14 and t(14;14)(q11;32) | TCL-1 (14q32.1) | T-CLL | IgH | ||
t(10;14)(q24;q32) | lyt-10 (10q24) | B lymphoma | IgH | ||
t(14;19)(q32;q13.1) | bcl3 (19q13.1) | B-CLL | IgH | ||
t(5;14)(q31;q32) | IL3 (5q31) | Pre-B-ALL | TCRβ | ||
t(7;9)(q34;q34.3) | tan1 (9q34.3) | T-ALL | TCRα | ||
t(1;7)(p34;q34) | lck (1p34) | T-ALL | TCRα | ||
t(X;14)(q28;11) | C6.1B (Xq28) | T-PLL |
Type | Affected gene | Protein domain | Fusion protein | Disease |
|---|---|---|---|---|
Gene fusions/hematopoietic tumors | ||||
inv 14(q11;q32) | TCRα (14q11) | TCR-Cα | VH-TCR-Cα | T/B-cell lymphoma |
VH (14q32) | lg VH | |||
t(9;22)(q34;q11) | CABL (9q34) | Tyrosine kinase | Serine + tyrosine kinase | CML/ALL |
bcr (22q11) | Serine kinase | |||
t(1;19)(q23;p13.3) | PBX1 (1q23) | HD | AD + HD | Pre-B-ALL |
E2A (19p13.3) | AD-bHLH | |||
t(17;19)(q22;p13) | HLF (17q22) | bZIP | AD + bZIP | Pro-B-ALL |
E2A (19p13) | AD-b-HLH | |||
t(15;17)(q21-q11-22) | PML (15q21) | Zinc finger | Zn-finger + RAR DNA | APL |
RARα (17q21) | Retinoic acid receptor-α | and ligand binding | ||
t(11;17)(q23;q21.1) | PLZF (11q23) | Zinc-finger | Zn-finger + RAR DNA | APL |
RARα (17q21) | Retinoic acid receptor-α | and ligand binding | ||
t(4;11)(q21;q23) | mll (11q23) | A-T hook/Zn-finger | A-T hook + Ser-pro | ALL/PreB-ALL/ANLL |
AF4 (4q21) | Ser-Pro rich | |||
t(9;11)(q21;q23) | AF9/MLLT3 (9p22) | (Ser-Pro rich) | A-T hook + (Ser-pro) | ALL/PreB-ALL/ANLL |
mll (11q23) | A-T hook/Zn-finger | |||
t(11;19)(q23;p13) | mll (11q23) | A-T hook/Zn-finger | A-T hook + Ser-pro | Pre-B-ALL/ |
ENL (19p13) | Ser-Pro rich | T-ALL/ANLL | ||
t(X;11)(q13;q23) | AFX1 (Xq13) | (Ser-Pro rich) | A-T hook + (Ser-pro) | T-ALL |
mll (11q23) | A-T hook/Zn-finger | |||
t(1;11)(p32;q23) | AF1P (1p32) | Eps-15 homolog | A-T hook + ? | ALL |
mll (11q23) | A-T hook/Zn-finger | |||
t(6;11)(q27;q23) | AF6 (6q27) | Myosin homolog | A-T hook + ? | ALL |
mll (11q23) | A-T hook/Zn-finger | |||
t(11;17)(q23;q21) | mll (11q23) | A-T hook/Zn-finger | A-T hook + leucine zipper | AML |
AF17 (17q21) | Cys-rich/leucine zipper | |||
t(8;21)(q22;q22) | eto/MTG8 (8q22) | Zn-finger | DNA binding + | AML |
aml1/CBFα (21q22) | DNA binding Zn-fingers | |||
Runt homology | ||||
t(3;21)(q26;q22) | evi-1 (3q26) | Zn-finger | DNA binding + | CML |
aml1 (21q22) | DNA binding | Zn-fingers | ||
t(3;21)(q26;q22) | EAP (3q26) | Sn Protein | DNA binding + | Myelodysplasia |
aml1 (21q22) | DNA binding | out-of-frame EAP | ||
t(16;21)(p11;q22) | FUS (16p11) | Gln-Ser-Tyr/Gly-rich/ | Gln-Ser-Tyr + | Myeloid |
RNA binding | DNA binding | |||
erg (21q22) | Ets-like DNA binding | |||
t(6;9)(p23;q34) | dek (6p23) | ? | ? + ZIP | AML |
can (9q34) | ZIP | |||
9;9? | set (9q34) | ? | ? + ZIP | AUL |
can (9q34) | ZIP | |||
t(4;16)(q26;p13) | IL2 (4q26) | IL2 | IL2/TM | T-lymphoma |
BMC (16p13.1) | ?/TM domain | |||
inv(2;2)(p13;p11.2-p14) | rel (2p13) | DNA binding-activator | DNA binding + ? | NHL |
NRG (2p11.2-p12) | ? | |||
inv(16)(p13;q22) | Myosin MYH11 (16p13) | DNA binding? | AML | |
CBFβ (16q22) | ||||
t(5;12)(q33;p13) | PDGFb (5q33) | Receptor kinase | Kinase + DNA binding | CMML |
TEL (12p13) | Ets-like DNA binding | |||
t(2;5)(p23;q35) | NPM (5q35) | Nucleolar phosphoprotein | N-terminus NPM | NHL |
ALK (2p23) | Tyrosine kinase | + kinase | ||
Gene fusions/solid tumors | ||||
inv10(q11.2;q21) | ret (10q11.2) | Tyrosine kinase | Unk + tyrosine kinase | Papillary thyroid |
D10S170 (q21) | Uncharacterized | Carcinoma | ||
t(11;22)(q24;q12) | fli1 (11q24) | Ets-like DNA binding | Gin-Ser-Tyr + DNA binding | Ewing’s sarcoma |
ews (22q12) | Gin-Ser-Tyr/Gly-rich/RNA binding | |||
t(21;22)(?;q12) | erg (21q22) | Ets-like DNA binding | Gin-Ser-Tyr + DNA binding | Ewing’s sarcoma |
ews (22q12) | Gin-Ser-Tyr/Gly-rich/RNA binding | |||
t(12;22)(q13;q12) | AFT1 (12q13) | bZIP | Gin-Ser-Try-bZIP | Melanoma |
ews (22q12) | Gin-Ser-Tyr/Gly-rich/RNA binding | |||
t(12;16)(q13;p11) | CHOP (12q13) | (DNA binding?)/ZIP | Gin-Ser-Tyr | Liposarcoma |
FUS (16p11) | Gin-Ser-Tyr/Gly-rich/RNA binding | |||
t(2;13)(q35;q14) | PAX3 (2q35) | Paired box/homeodomain | + (DNA binding?)/ZIP | Rhabdomyosarcoma |
FKHR (13q14) | Forkhead domain | |||
t(X;18)(p11.2;q11.2) | SYT (18q11.2) | None identified | PB/HD + DNA binding | Synovial sarcoma |
SSX (Xp11.2) | None identified |
Consequent to rapid advances in cytogenetic resolution techniques, Rowley in 1973 [14] found that the Philadelphia chromosome actually resulted from a reciprocal translocation involving the long arms of chromosomes 9 (9q34) and 22 (22q11). Analysis of the affected region on chromosome 9 revealed a proto-oncogene, c-abl [15], which when translocated to chromosome 22 generates a fusion gene. The c-abl proto-oncogene has 11 exons that encode for a 145 kDa protein with tyrosine kinase activity. The chromosomal breakpoint within the c-abl gene consistently involves one of two alternatively spliced exons. Breakpoints along the functional gene on chromosome 22 are clustered near the center in a 6 kb region termed the breakpoint cluster region (bcr). Upon translocation, nearly the entire c-abl proto-oncogene is placed under bcr promoter activity. Transcription and splicing yield a long mRNA transcript encoding a chimeric 210 kDa protein that expresses increased tyrosine kinase activity, likely because it is less responsive to normal regulatory elements (Fig. 7.1). A similar translocation exists in some acute lymphoblastic leukemias, although the breakpoint occurs further upstream in the bcr gene which results in a smaller chimeric protein (190 kDa), which has also been shown to have increased tyrosine kinase activity [16–20].
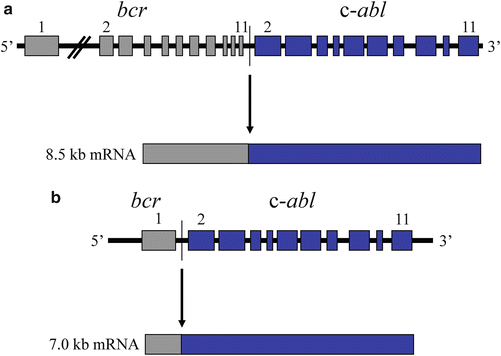
Fig. 7.1
bcr-abl translocation in chronic myelogenous leukemia. The c-abl proto-oncogene on chromosome 9 is translocated to the breakpoint cluster region (bcr) of chromosome 22. The result is a novel tyrosine kinase which functions independently of normal regulatory elements. (a) The t(9;22) that is commonly observed in chronic myelogenous leukemia (CML). (b) The t(9;22) that is commonly observed in acute lymphocytic leukemia.
Translocation leading to dysregulation of c-myc . Investigation into the role of the c-myc proto-oncogene in neoplastic transformation led to development of a model of proto-oncogene activation based upon insertional mutagenesis. This mechanism of proto-oncogene activation emerged from studies of acutely transforming retroviruses and weakly oncogenic or non-transforming retroviruses. The primary differences between these two classes of retrovirus reflect the amount of time necessary for induction of tumors after infection of cells and the genomic content of their proviral DNA. Acutely transforming retroviruses have oncogenes incorporated into their genome while non-transforming retroviruses do not. Thus, the transformation potential of weakly oncogenic and non-transforming retroviruses depend on insertion adjacent to a cellular proto-oncogene. Although retroviruses insert randomly, in independently derived tumors retroviral sequences were found incorporated into similar chromosomal locations in the host genomic DNA. The site of insertion then became the focus of attention, and cellular homologs of known retroviral oncogenes and their surrounding sequences were studied intensely. Finally, Hayward and Astrin demonstrated that non-acutely transforming retroviruses insert adjacent to and cause activation of the cellular proto-oncogene c-myc [21]. Insertional mutagenesis is based on the ability of proviral DNA to insert into host genomic DNA and cause either activation or inactivation of host genes, independent of expression of retroviral genes (as in the case of acutely transforming viruses). In the case of insertional activation of cellular genes, the proviral DNA may provide a promoter or enhancer for the cellular gene, resulting in an alteration in the normal regulation and expression pattern of the affected gene.
In the early 1980s data on c-myc activation by non-acutely transforming retroviruses in chicken lymphomas merged with data accumulating on translocations in Burkitt’s lymphoma, a high grade B lymphocyte neoplasm. It was reasoned that if proviral sequences were capable of altering host cellular gene expression to cause tumors, chromosomal alterations that juxtapose promoter or enhancing sequences and cellular proto-oncogenes (through chromosomal translocation) were likely to promote neoplastic transformation. The best studied translocations at the time involved those of Burkitt’s lymphoma, in which a portion of the long arm of chromosome 8 is consistently translocated to either chromosome 14, 2, or 22, adjacent to the loci for immunoglobulin heavy chain, k light chain, and λ light chain, respectively. The immunoglobulin loci on chromosomes 14, 2, and 22 were postulated to be good partner candidates to be coupled with and cause activation of a proto-oncogene that was suspected to reside on chromosome 8. Tumor DNA was directly probed for c-myc sequences. The gene was detected on chromosome 8, and found to be translocated to chromosomes 14, 2, and 22 in Burkitt’s lymphoma and in some plasmacytomas. In plasmacytomas, a form of the c-myc proto-oncogene lacking the untranslated exon 1 is involved in the chromosomal translocation (Fig. 7.2). The breakpoints within the c-myc gene are more varied in Burkitt’s lymphoma. Nonetheless, the translocation results in abnormal (constitutive) expression of a c-myc coding sequence identical to its normal allele in both types of tumor. This observation strongly suggested the chromosome translocation-mediated activation of the c-myc proto-oncogene as a causal event in human tumorigenesis [4, 22–25].
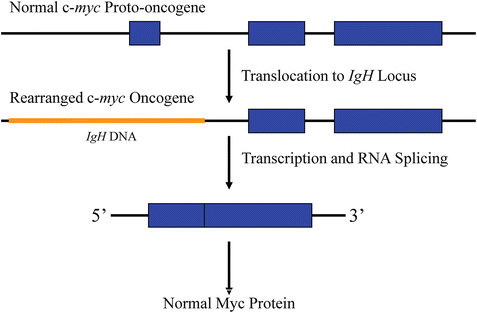
Fig. 7.2
Consequences of the t(8;14) chromosomal translocation. The c-myc gene of chromosome 8 is normally not expressed in differentiated B cells. After translocation, it comes under the control of either a cryptic promoter in intron 1 or an enhancer from the immunoglobulin locus of chromosome 14 (IgH), leading to constitutive expression of the normal c-myc protein (exon 1 is noncoding). IgH gene sequences are depicted in orange and c-myc gene sequences are depicted in blue.
7.2.3.2 Proto-oncogene Activation Through Gene Amplification
Activation of cellular proto-oncogenes can occur as a consequence of DNA amplification resulting in overexpression of the amplified proto-oncogene, which confers a proliferative advantage to affected cells. Amplified gene segments can be discerned cytogenetically as double minute chromosomes (DMs) and homogeneously staining regions (HSRs). Proto-oncogenes (and other genetic loci) are amplified by repeated DNA replication events that can result in an abnormal homogeneous staining pattern in a karyotypic analysis of affected cells, rather than the familiar chromosomal staining pattern in R-banded or G-banded chromosome spreads. Instead, homogeneously staining regions appear as abnormally extended R-bands or G-bands (Fig. 7.3). The tandem arrays of amplified DNA forming homogeneously staining regions may be excised from the chromosome to form double minutes, which are small chromosomal structures lacking centromeres that do not replicate during cell division. Double minutes may integrate into other chromosomes to create additional stable HSRs able to propagate upon cell division [26, 27].
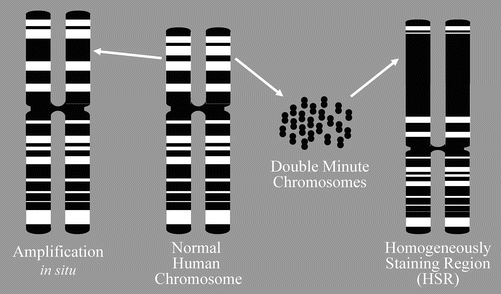
Fig. 7.3
DNA amplification . When DNA is amplified by repeated DNA replication events, the results can sometimes be seen cytogenetically as homogeneously staining regions (HSRs) or double minutes. Double minutes represent the extrachromosomal manifestation of HSRs. Double minutes can insert into any chromosome (the one they are derived form or a different chromosome).
In the same way that investigation of the c-myc proto-oncogene formed the underpinnings of our current understanding of proto-oncogene activation by means of chromosomal translocation, studies of the c-myc proto-oncogene led to the unraveling of proto-oncogene activation through gene amplification in human neoplasms [28]. DNA amplification represents one mechanism leading to drug resistance in mammalian cells [29]. Through direct probing for c-myc it was discovered that double minutes and homogeneously staining regions contained amplified copies of the oncogene in human colon carcinoma cells [30]. The c-myc gene has been shown to be amplified and overexpressed in a number of human neoplasms supporting the role of DNA amplification as a major mechanism for cellular proto-oncogene activation in neoplastic transformation.
Although the precise mechanism for gene amplification has not been entirely determined, the role amplification plays in cellular transformation in human malignancies is clear, particularly from studies of neuroblastomas and studies involving neoplastic transformation of cells in vitro [31]. Neuroblastoma is one of the most common childhood extracranial solid tumors accounting for approximately 15 % of all childhood cancer deaths [32]. Neuroblastomas exhibit DMs and HSRs that hybridize with probes to the c-myc gene. The hybridizing sequences were determined to be related to but distinct from c-myc and was designated N-myc [33]. The N-myc gene is transcribed at higher levels in neuroblastomas that demonstrate gene amplification. N-myc amplification is now a major prognostic determinant in neuroblastomas, with high levels of transcription from either a single copy or more commonly from increased gene copy number in the form of DMs or HSRs correlating with poor patient survival [34]. The demonstration of the link between high N-myc expression and poor clinical prognosis, and its demonstrated ability to cause neoplastic transformation in cell culture provides strong evidence for the importance of gene amplification in the activation of cellular proto-oncogenes. Table 7.3 lists other cellular proto-oncogenes that have been shown to be amplified in human neoplasms.
Table 7.3




Oncogene amplification in human tumors
Related posts:




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
