AMENORRHEA
DEFINITION
Amenorrhea is generally defined as the absence of menstruation for 3 or more months in women with past menses or a failure to menstruate by girls 16 years of age who have never menstruated. Amenorrhea is merely a sign; it may suggest several disorders involving any of several organ systems. If the genital outflow tract is intact, amenorrhea indicates failure of the hypothalamicpituitarygonadal axis to interact to induce the cyclic changes in the endometrium that normally cause menses. Amenorrhea may be the result of an abnormality at any level of the reproductive tract.
Traditionally, amenorrhea is regarded as primary in women who have never menstruated and as secondary in women who have menstruated previously. Because such categorization may lead to diagnostic omission, whether the amenorrhea is primary or secondary should not be a major factor in the evaluation of an amenorrheic woman. Similarly, use of the term “postpill” amenorrhea to refer to women who fail to resume menses within 3 months of discontinuing oral contraceptives conveys nothing about the cause. Women who have fewer than 9 menstrual periods per calendar year should be evaluated identically to those with amenorrhea.
CLINICAL EVALUATION
Most important to the clinical evaluation are the history and physical examination, with special attention to the possible effects of alterations in hormonal secretion on pubertal development. In general, the clinician should view the patient as a bioassay subject in whom gonadal steroids lead normally to the development of secondary sex characteristics. Breast development indicates exposure to estrogens. The presence of pubic and axillary hair indicates exposure to androgens. Any abnormality of the outflow tract should be eliminated by physical examination.
Patients should be questioned regarding the timing of pubertal milestones, and any abnormalities of growth and development should be pursued (see Chap. 91 and Chap. 92).
Patients also should be asked about dietary and exercise habits; other aspects of lifestyle, environmental, and psychological stresses; and any family history of amenorrhea or genetic anomalies. It is also important to search for any signs of increased levels of androgen, including acne, hirsutism (i.e., increased sexually stimulated terminal hair; see Chap. 101), and even virilization, such as increased masculine and decreased feminine secondary sexual characteristics, including hirsutism, temporal balding, deepening of the voice, increased muscle mass, clitoromegaly, decreased breast size, and vaginal atrophy. Any history of galactorrhea (i.e., nonpuerperal secretion of milk) should be elicited.
Patients also should be asked about dietary and exercise habits; other aspects of lifestyle, environmental, and psychological stresses; and any family history of amenorrhea or genetic anomalies. It is also important to search for any signs of increased levels of androgen, including acne, hirsutism (i.e., increased sexually stimulated terminal hair; see Chap. 101), and even virilization, such as increased masculine and decreased feminine secondary sexual characteristics, including hirsutism, temporal balding, deepening of the voice, increased muscle mass, clitoromegaly, decreased breast size, and vaginal atrophy. Any history of galactorrhea (i.e., nonpuerperal secretion of milk) should be elicited.
Body dimensions and habitus, the distribution and extent of body hair, breast development and secretions, and the external and internal genitalia should be carefully evaluated. Because disorders of sexual development and reproduction frequently are manifested by changes in habitus, it is important to consider the patient’s overall appearance. In normal adults, the arm span is similar to the height; in hypogonadal individuals, the arm span typically exceeds the height by 5 cm or more. In congenital hypothyroidism, the extremities are significantly shorter than in normal individuals.
The distribution and quantity of body hair should be evaluated, especially with reference to the family history. Hypertrichosis, or the excessive growth of terminal hair on the back and extremities, is almost invariably familial and must be differentiated from true hirsutism. Hypertrichosis is common in women of Mediterranean ancestry, but any facial hair growth in Asian and American Indian women demands evaluation. Although several semiquantitative methods of scoring hirsutism have been developed, it is perhaps most practical to grade facial hirsutism only (because this usually is of most concern to the patient) from 0 to 4+, assigning one point each for excess chin, upper lip, or sideburn hair, and 4+ for a complete beard.1 For documentation, there is no substitute for photographs.
Breast development should be staged according to the method of Tanner2 (see Chap. 91). The breasts should be examined for any secretion by applying pressure to all sections of the breast, beginning lateral to the nipple and working toward the nipple while the patient is seated. Secretions should be examined microscopically as a wet mount for the presence of thick-walled, perfectly round fat globules of various sizes, establishing that the discharge is milk (Fig. 96-1).
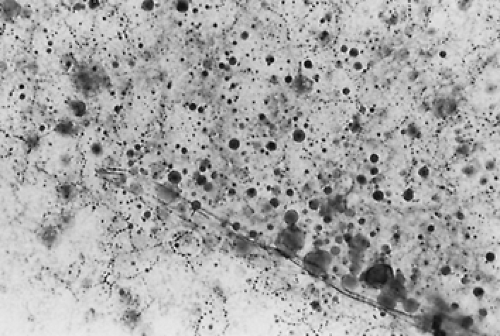 FIGURE 96-1. Perfectly round, thick-walled fat globules of various sizes are characteristic of galactorrhea when the breast secretion is viewed as a wet preparation under the microscope (original magnification, × 88). For photography, the oil red O stain was added to the specimen, accounting for the dark character of the fat droplets. |
The female genitalia are the most sensitive indicators of hormonal status. The Tanner stage of pubic hair development should be recorded.2 The extent of any virilization present indicates the stage in development when exposure to androgens occurred; in general, the sensitivity of the genitalia to androgens decreases with time from the early stages of fetal development to adulthood. The most significant changes, including fusion of the labia and enlargement of the clitoris with or without formation of a penile urethra, are found in women exposed to excess androgens during the first few months of fetal development, as in congenital adrenal hyperplasia (see Chap. 77). The development of significant clitoromegaly in an adult requires marked androgenic stimulation and strongly suggests the presence of an androgen-secreting neoplasm (see Chap. 102). The glans clitoris is definitely enlarged if it is 1 cm or more in diameter. A clitoral index, defined as the product of the sagittal and transverse diameters of the glans at the base, greater than 35 mm2 falls outside the 95% confidence interval.3 Under the influence of estrogen, the labia minora develop at puberty.
Examination of the internal genitalia should reveal any overt anomalies of müllerian duct derivatives, including imperforate hymen, vaginal and uterine aplasia, and vaginal septum (see Chap. 90). Obstruction to the escape of menstrual blood can cause hematocolpos (i.e., collection of blood in the vagina) and hematometra (i.e., distention of the uterus with blood). Although a bulging perineum and a pelvic mass are typically found on examination, differentiating vaginal agenesis from a vaginal septum or an imperforate hymen may be difficult. In all of these cases, the normal development of the external genitalia and of other secondary sex characteristics indicates normal ovarian function. The occurrence of intermittent abdominal pain suggests intra-abdominal bleeding. Müllerian dysgenesis (i.e., Rokitansky-Küster-Hauser syndrome) may be accompanied by bony abnormalities of the lumbar spine (e.g., spina bifida occulta), renal anomalies, and disorders of the eighth cranial nerve.4
If there is asynchronous pubertal development with significant breast development in the absence of much pubic and axillary hair, androgen insensitivity (i.e., 46,XY male pseudohermaphroditism) must be excluded. These disorders, including complete testicular feminization, are generally inherited as X-linked recessive or sex-linked autosomal-dominant traits. Complete virilization does not occur despite the presence of testes located inguinally or intraabdominally. Patients have a typical female habitus with normal female external genitalia, but breasts develop only to Tanner stage 3, and the vagina is absent or ends blindly (see Chap. 90).
Outflow tract obstruction associated with a normal uterus should be treated surgically to prevent tubal damage from intraabdominal menstruation. Individuals with testicular feminization should be reared as females and treated with an estrogen and a progestin after surgical removal of the testes. The testes should be removed because of the risk of malignancy. Girls lacking a vagina may undergo vaginoplasty (i.e., McIndoe procedure) when regular sexual activity is anticipated.5 In motivated individuals, a vagina can also be created gradually by the daily use of dilators of increasing size (i.e., Frank nonoperative method).6
For individuals with a normal genital tract, visual inspection of the quality of the vaginal mucosa and of the cervical mucus is important because the two are sensitive to estrogen. In response to this hormone, the vaginal mucosa is transformed at puberty from a tissue with a shiny, smooth, bright red appearance to a dull, gray-pink, rugated surface. The cervical mucus increases in quantity and elasticity (i.e., spinnbarkeit) when estrogen is present. Pelvic examination may also reveal pelvic pathologic processes, including neoplasms.
The history and physical examination can differentiate several causes of amenorrhea in women of reproductive age, including disorders of sexual differentiation (e.g., distal genital tract obstruction such as müllerian agenesis and dysgenesis, gonadal dysgenesis, ambiguity of external genitalia as in male and female pseudohermaphroditism); other peripheral causes (e.g., pregnancy, gestational trophoblastic disease, amenorrhea traumatica as in Asherman syndrome); and chronic anovulation or ovarian failure (e.g., hypothalamic-pituitary dysfunction,
inappropriate feedback because of polycystic ovarian syndrome, adrenal or thyroid dysfunction, abnormal prolactin secretion, premature ovarian failure).
inappropriate feedback because of polycystic ovarian syndrome, adrenal or thyroid dysfunction, abnormal prolactin secretion, premature ovarian failure).
Any sexual ambiguity indicates the need for chromosomal karyotyping and the measurement of serum 17-hydroxy-progesterone to rule out 21-hydroxylase deficiency (e.g., congenital adrenal hyperplasia; see Chap. 77). Pregnancy and gestational trophoblastic disease may be confirmed by determining if circulating levels of human chorionic gonadotropin (hCG) are elevated. The existence of intrauterine synechiae or adhesions (i.e., Asherman syndrome) must be suspected in women who develop oligomenorrhea or amenorrhea after curettage or endometritis; tuberculous endometritis may also lead to this disorder.7 The diagnosis can be made by performing hysterosalpingography or hysteroscopy. Hysteroscopic lysis of adhesions is effective in treating Asherman syndrome in more than 80% of affected individuals.
Unless serum follicle-stimulating hormone (FSH) levels are measured, it is frequently impossible to differentiate individuals with chronic anovulation, in whom hypothalamic–pituitary–ovarian function is disrupted, from those patients with ovarian failure in whom the ovaries are generally devoid of oocytes. However, it should be possible to form strong clinical impressions about the cause of the amenorrhea.
To determine with certainty whether the outflow tract is intact and to evaluate the levels of endogenous estrogen, exogenous progestin, in the form of progesterone in oil (100–200 mg given intramuscularly) or medroxyprogesterone acetate (5–10 mg taken orally each day for 5 to 10 days), can be administered. Any genital bleeding within 10 days of the completion of progestin administration makes the diagnosis of Asherman syndrome unlikely (although still possible) and suggests the presence of chronic anovulation rather than hypothalamic-pituitary or ovarian failure. If the patient does not bleed in response to the progestin, an estrogen and a progestin together (e.g., 2.5 mg of oral conjugated estrogen daily for 25 days with 5 to 10 mg of oral medroxyprogesterone acetate or 200 mg of micronized progesterone also given for the last 10 days) should produce bleeding if the endometrium is normal. Withdrawal bleeding in response to progestin does not exclude the diagnosis of hypergonadotropic amenorrhea, associated with ovarian failure.
LABORATORY EVALUATION
After appropriate clinical evaluation, measurements of basal serum levels of FSH, prolactin, and thyroid-stimulating hormone (TSH) are indicated in all amenorrheic women to confirm the clinical impression (Fig. 96-2). Whenever the basal prolactin level is elevated (generally >20 ng/mL) on initial testing, the measurement should be repeated, because prolactin levels are increased by a number of nonspecific stimuli, including stress, sleep, and food ingestion. If thyroid function is normal and prolactin levels are elevated, further evaluation is warranted to rule out a pituitary tumor and other causes (see Chap. 13). Basal prolactin concentrations should be determined in all amenorrheic women, not just in those with galactorrhea, because prolactin levels are elevated in more than one-third of all amenorrheic women.8
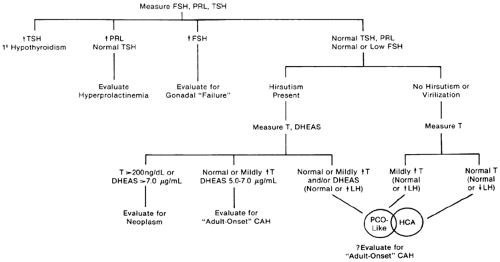 FIGURE 96-2. Flow diagram for the laboratory evaluation of amenorrhea. Such a scheme must be considered as an adjunct to the clinical evaluation of the patient. (CAH, congenital adrenal hyperplasia; DHEAS, dehydroepiandrosterone sulfate; FSH, follicle-stimulating hormone; HCA, hypothalamic chronic anovulation; PCO, polycystic ovarian syndrome; PRL, prolactin; T, thyroxine; TSH, thyroid-stimulating hormone.) (Reprinted from Rebar RW. The ovaries. In: Smith LH Jr, ed. Cecil textbook of medicine, 18th ed. Philadelphia: WB Saunders, 1992:1367.) |
Increased serum TSH levels (generally >5 μU/mL utilizing sensitive assays) with or without increased levels of prolactin indicate primary hypothyroidism (see Chap. 15 and Chap. 45). The increased secretion of thyrotropin-releasing hormone (TRH) in this disorder stimulates increased secretion of prolactin and TSH in some affected women.
High serum FSH levels (>30 mIU/mL in most laboratories) imply ovarian failure. Chromosomal evaluation is indicated in all women with increased serum FSH levels who are younger than 30 years of age when the amenorrhea begins, because a number of karyotypic abnormalities have been identified in such women. Gonadectomy is indicated in any such individual who has a portion of a Y chromosome because of the malignant potential of the gonads.9
If prolactin, TSH, and FSH levels are normal or low, further evaluation is based on the clinical presentation. Circulating thyroid hormone levels should be determined if there is any suggestion of thyroid dysfunction. Serum total testosterone levels should be determined whether or not the patient is hirsute; not all hyperandrogenic women are hirsute because of relative insensitivity of the hair follicles to androgens in some women. Although slightly increased levels of serum testosterone and perhaps of dehydroepiandrosterone sulfate (DHEAS) suggest polycystic ovarian syndrome (PCO), androgen levels occasionally are not elevated in PCO, because of alterations in the metabolic clearance rates of androgens and in sex-hormone-binding-globulin (SHBG) concentrations.10 Circulating levels of luteinizing hormone (LH) may also aid in differentiating PCO from hypothalamic-pituitary dysfunction or failure. LH levels often are increased in PCO such that the ratio of LH to FSH is increased, but this too is not always so.11 However, LH and FSH levels are normal or slightly reduced in women with hypothalamic-pituitary dysfunction.12
There is some overlap between women with PCO-like disorders and those with hypothalamic-pituitary dysfunction. In an effort not to miss a serious cause of amenorrhea, some radiographic assessment of the region of the sella turcica is indicated in all amenorrheic women in whom LH and FSH levels are low (generally <10 mIU/mL) to exclude a pituitary or hypothalamic
neoplasm. In addition to testosterone, serum DHEAS levels should be measured in women with evidence of hirsutism or virilization. Serum total testosterone levels of 200 ng/mL or greater should prompt an investigation for an androgen-producing neoplasm, most likely of ovarian origin. Serum DHEAS levels above 7.0 μg/dL should prompt an investigation for an adrenal neoplasm. Adultonset or cryptic congenital adrenal hyperplasia (see Chap. 77) should be considered in women with severe hirsutism since puberty, a strong family history of hirsutism, stature shorter than expected compared with other family members, or serum DHEAS levels of 5.0 μg/mL or greater.
neoplasm. In addition to testosterone, serum DHEAS levels should be measured in women with evidence of hirsutism or virilization. Serum total testosterone levels of 200 ng/mL or greater should prompt an investigation for an androgen-producing neoplasm, most likely of ovarian origin. Serum DHEAS levels above 7.0 μg/dL should prompt an investigation for an adrenal neoplasm. Adultonset or cryptic congenital adrenal hyperplasia (see Chap. 77) should be considered in women with severe hirsutism since puberty, a strong family history of hirsutism, stature shorter than expected compared with other family members, or serum DHEAS levels of 5.0 μg/mL or greater.
HYPERGONADOTROPIC AMENORRHEA (PRIMARY HYPOGONADISM)
It is frequently impossible to diagnose hypergonadotropic amenorrhea, also called presumptive ovarian failure, without the measurement of basal serum gonadotropin levels. This is especially true because ovarian failure may occur at any time from embryonic development onward. The ovaries normally fail at the time of menopause, when virtually no viable oocytes remain. The premature loss of oocytes before age 40 results in premature ovarian failure. From what is known about follicular development and atresia, it appears that premature ovarian failure arises from abnormalities in the recruitment and selection of oocytes13 (see Chap. 94). The follicles may undergo atresia at an accelerated rate or a smaller than normal pool may undergo atresia at the normal rate to yield early oocyte depletion. FSH must be involved, because it is the principal hormonal regulator of folliculogenesis. Circulating gonadotropin levels increase whenever ovarian failure occurs because of decreased negative estrogen feedback to the hypothalamic-pituitary unit. Gonadotropin levels sometimes increase even in the presence of viable oocytes, but the explanation for such increases is unclear. In principle, young women with hypergonadotropic amenorrhea must be considered potentially, if only rarely, fertile.
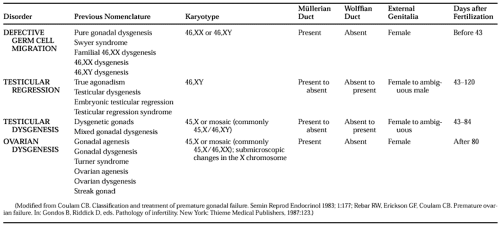
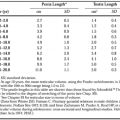
It is possible to identify disorders causing gonadal failure before birth that arise from gonadal abnormalities (Table 96-1; see Chap. 90). This classification emphasizes the need to consider genetic abnormalities in all young women with hypergonadotropic amenorrhea. In ovarian dysgenesis, the normal complement of oocytes is present at 20 weeks of fetal age, but atresia accelerates until birth, and some affected girls may undergo sexual development, ovulate, and even conceive if some oocytes remain.14,15 The classification in Table 96-1 is not ideal but should be viewed as an artificial categorization of a continuum. Excluding these disorders of gonadal development, premature ovarian failure still consists of several distinct disorders (Table 96-2).
 |
 |
TYPES OF PREMATURE OVARIAN FAILURE
Inherited characteristics are important in the development of premature ovarian failure. Premature loss of oocytes could result from a reduced germ cell endowment in utero, accelerated atresia, or failure of all germ cells to migrate to the genital ridges in early development (see Chap. 90). There may be marked differences in oocyte endowment and rates of follicular atresia among women.16,17 The cause of the ovarian failure that may accompany myotonia dystrophica is unknown but may involve decreased germ cell number or accelerated atresia.18 An excess of X chromosomes may also be associated with decreased germ cell numbers or accelerated atresia.19 The familial occurrence of premature ovarian failure with vertical transmission has been described, suggesting autosomal-dominant, sex-linked inheritance in some cases.20 This observation has significant implications for the reproductive counseling of affected women who had children before developing ovarian failure.
At least two separate inherited enzymatic defects may also be associated with premature ovarian failure. Girls with 17α-hydroxylase deficiency who survive to their teens present with sexual infantilism; primary amenorrhea; increased circulating levels of LH, FSH, deoxycorticosterone, and progesterone; and hypertension with hypokalemic alkalosis21,22 and 23 (see Chap. 77). Ovarian biopsy has revealed no evidence of orderly follicular maturation but instead has demonstrated numerous large cysts and primordial follicles. Presumably, the enzyme deficiency does not permit normal follicular development. The startling observation that normal follicular growth and development with successful fertilization in vitro can be achieved with exogenous gonadotropins in individuals with 17α-hydroxylase deficiency
raises significant questions about why there is no follicular development in affected girls24 (see Chap. 95). Girls with galactosemia, a disorder in which galactose-1-phosphate uridyltransferase activity is decreased and that is characterized by mental retardation, cataracts, hepatosplenomegaly, and renal tubular dysfunction, may also develop premature ovarian failure with hypergonadotropinism even when a galactose-restricted diet is introduced early in infancy.25
raises significant questions about why there is no follicular development in affected girls24 (see Chap. 95). Girls with galactosemia, a disorder in which galactose-1-phosphate uridyltransferase activity is decreased and that is characterized by mental retardation, cataracts, hepatosplenomegaly, and renal tubular dysfunction, may also develop premature ovarian failure with hypergonadotropinism even when a galactose-restricted diet is introduced early in infancy.25
At least one form of premature ovarian failure is caused by mutations of the FSH receptor (FSHR). (Because some clinicians regard this disorder as another form of XX gonadal dysgenesis, this disorder is also discussed in Chap. 90.) Because affected individuals present with primary or secondary amenorrhea and elevated levels of FSH and ovarian follicles may be detected during transvaginal ultrasound examination, it may be argued that the disorder is more appropriately included here.
One specific mutation on chromosome 2p (C566T:alanine to valine) in exon 7 of the FSHR has been identified in several Finnish families,26,27 but the mutation must be very rare outside of Finland because it has not been detected in some other populations.28,29
The “resistant ovary” syndrome may be the result of a gonadotropin postreceptor defect. As originally described, the Savage syndrome (named after the first patient described) consisted of young amenorrheic women with elevated peripheral gonadotropin concentrations, normal but immature follicles in the ovaries on biopsy, 46,XX karyotype with no evidence of mosaicism, complete sexual development, and hyposensitivity (i.e., “resistance”) to exogenous gonadotropin stimulation.30 Although the pathogenesis of this disorder remains obscure, it may be reasonable to consider affected individuals distinct from those with autoimmune disorders.
Premature ovarian failure may be associated with a number of autoimmune disorders.13 The most common association may be with thyroiditis. Ovarian failure occurs commonly in women with polyglandular failure, including hypoparathyroidism, hypoadrenalism, and mucocutaneous candidiasis31 (see Chap. 197). The heterogeneous nature of this disorder is suggested by the many different endocrinopathies that may be associated with premature ovarian failure. Autoimmune ovarian failure may occur independently of any other autoimmune disorder.30a Although the cause of autoimmune ovarian failure is unknown, circulating antibodies to ovarian tissue have been detected in many affected women, and FSH receptor antibodies have been detected in two women with myasthenia gravis and hypergonadotropic amenorrhea.32 Immunoglobulins that block the trophic actions of FSH but not LH have also been reported.33
The thymus gland influences reproductive function34 (see Chap. 193). Congenitally athymic girls have ovaries devoid of oocytes.35
Irradiation and an increasing number of chemotherapeutic (especially alkylating) agents used to treat various malignancies are causes of premature ovarian failure36,37 and 38 (see Chap. 226). Inexplicably, both of these modalities have been associated with “reversible” ovarian failure. Ovulation and cyclic menses return in some individuals after prolonged intervals of hypergonadotropic amenorrhea associated with signs and symptoms of profound hypoestrogenism. Preliminary studies suggest that gonadotropin-releasing hormone analogs (but not oral contraceptive agents) may provide some protection from ovarian damage.39 Rarely, the mumps virus can affect the ovaries and cause ovarian failure.40
Based on epidemiologic studies, there now appears to be an inverse dose-response relationship between the number of cigarettes smoked per day and the age at menopause. At any given age between 44 and 53 years, a woman who smokes one pack per day is more likely to have undergone menopause than a woman who smokes one-half pack per day or less41 (see Chap. 234). However, because cigarette smoking is so prevalent and premature ovarian failure so uncommon, it is difficult to believe that smoking plays any significant role in inducing early ovarian failure. Moreover, because relatively few cases of premature ovarian failure appear to be the result of environmental factors, a cause distinct from an environmental cause should be sought in any affected woman without a history of systemic disease, radiation therapy, or chemotherapy.
DIAGNOSIS AND THERAPY OF PREMATURE OVARIAN FAILURE
Individuals with premature ovarian failure warrant thorough evaluation to eliminate potentially treatable causes and to identify associated disorders that may require therapy. Chromosomal studies should be performed in affected women younger than 30 years of age to identify those with gonadal dysgenesis who have no signs of Turner syndrome. Surgical removal of the gonads is indicated in any individual in whom a Y chromosome is identified. A few simple laboratory studies can rule out endocrine disorders, such as thyroid disease, hypoparathyroidism, hypoadrenalism, and diabetes mellitus. Screening tests to eliminate autoimmune dysfunction are indicated as well. Measurement of circulating LH, FSH, and estradiol concentrations on more than one occasion may help to determine if any functional oocytes remain in the ovary. If the estradiol concentration is >50 pg/mL or if the LH level is significantly greater than the FSH level (in terms of mIU/mL) in any sample, the probability of viable oocytes is considerable. Irregular uterine bleeding, as an indication of estrogen stimulation, also provides good evidence of remaining functional ovarian follicles. It is not uncommon to
identify women with intermittent menstruation, hypoestrogenism, and hypergonadotropinism. Because a number of pregnancies have occurred after biopsy of ovaries devoid of oocytes, ovarian biopsy cannot be recommended for affected women.
identify women with intermittent menstruation, hypoestrogenism, and hypergonadotropinism. Because a number of pregnancies have occurred after biopsy of ovaries devoid of oocytes, ovarian biopsy cannot be recommended for affected women.
Even in women with intermittent ovarian failure, estrogen replacement is appropriate to prevent the accelerated bone loss that occurs in affected women.42 The estrogen should always be given sequentially with a progestin to prevent endometrial hyperplasia (see Chap. 100). Because women with ovarian failure may conceive while on estrogen therapy (including combined oral contraceptive agents), affected women should be counseled appropriately and cautioned to have a pregnancy test if withdrawal bleeding does not occur or if signs and symptoms develop that are suggestive of pregnancy. Despite these considerations, probably no other contraceptive agent is required for those women who do not wish pregnancy but who are sexually active, because pregnancy occurs in far less than 10%.13 Although rare pregnancies in women with premature ovarian failure have occurred after ovulation induction with human menopausal and chorionic gonadotropins, the low likelihood should lead the physician to discourage patients from selecting such therapy. Hormone replacement treatment to mimic the normal menstrual cycle, with oocyte donation for embryo transfer, may provide the greatest possibility for pregnancy in women desiring pregnancy.43,44
CHRONIC ANOVULATION
Chronic anovulation may be viewed as a steady state in which the monthly rhythms associated with ovulation are not functional. Although amenorrhea is common, irregular menses and oligomenorrhea may occur as well. Chronic anovulation further implies that viable oocytes remain in the ovary and that ovulation can be induced with appropriate therapy.
Chronic anovulation is the most common endocrine cause of oligomenorrhea or amenorrhea in women of reproductive age (Table 96-3). Appropriate management requires determination of the cause of the anovulation. However, anovulation can be interrupted transiently by nonspecific induction of ovulation in most affected women.
 |
CHRONIC ANOVULATION OF CENTRAL ORIGIN
Hypothalamic Chronic Anovulation.
Hypothalamic chronic anovulation may be defined as anovulation in which dysfunction of hypothalamic signals to the pituitary gland causes failure to ovulate. It remains unclear whether the primary abnormality is always present within the hypothalamus or sometimes occurs as a result of altered inputs to the hypothalamus. The term is used to refer to women who may be affected with suprahypothalamic or hypothalamic chronic anovulation. Although isolated gonadotropin deficiency frequently is caused by hypothalamic dysfunction, it is preferable to consider such individuals separately. However, it may be virtually impossible to differentiate partial forms of isolated gonadotropin deficiency from hypothalamic chronic anovulation.
Some reports have documented an increased incidence of amenorrhea in women who exercise strenuously, diet excessively, or are exposed to severe emotional or physical stresses of any kind45,46 and 47 (see Chap. 128). Such amenorrheic persons fall into this group of women considered as having hypothalamic chronic anovulation, which is sometimes called functional amenorrhea. The diagnosis of hypothalamic chronic anovulation is suggested by the abrupt cessation of menses in women younger than 30 years of age who have no clinically evident anatomic abnormalities of the hypothalamic–pituitary–ovarian axis or any other endocrine abnormalities. The term hypothalamic amenorrhea was first proposed by Klinefelter and colleagues in 1943 for anovulation in which hypothalamic dysfunction is thought to interfere with the pituitary secretion of gonadotropin.48
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


