Introduction
Alzheimer’s disease (AD) is the most common form of dementia in older adults.1 AD symptoms include a loss of memory, impaired judgment and decision-making capacity, a decline in the ability to perform activities of daily living (ADLs), changes in behaviour, mood and personality and increasing dependence on caregivers with expanding degrees of burden and stress.
Alois Alzheimer described the first case in 1906, characterizing a 51-year-old woman who presented with delusions of spousal infidelity, memory and language problems. After her death, using recently developed silver staining techniques, Alzheimer described numerous senile plaques and neurofibrillary tangles characteristic of the disease.2 For the next 60 years, AD was thought to be an infrequent pre-senile cause of dementia until it was recognized that the clinical symptoms and course and neuropathological findings of disease in individuals younger than 65 years was identical with those found in older adults.
The Alzheimer Association estimates that there are over 5 million people in the USA with AD and that within a generation the number of AD patients will exceed 15 million people.3 In the USA, AD is the fifth leading cause of death, after cardiovascular disease, cancer, cerebrovascular disease and bronchopulmonary diseases. AD is the third most expensive disease after cardiovascular disease and cancer in terms of total dollars with an annual total (direct and indirect) cost of approximately $172 billion. While the costs of AD increase across the stages of severity, AD patients currently fill nearly half of all nursing home beds. This economic impact will continue to grow as the US population continues to age and the number of AD patients increases. At the time of writing, the cumulative costs of care for people with AD from 2010 to 2050 in the USA will exceed $20 trillion.3 At present, only symptomatic therapies are available that provide cognitive, functional and behavioural benefits but do not alter the disease course.
Neuropathology
Macroscopically, AD is characterized by cortical atrophy and ventricular dilatation (Figure 73.1a). Volume is most noticeable on coronal sections with shrinkage of medial temporal lobe structures including hippocampus. The characteristic pathological changes in the AD brain are the accumulation of amyloid β-protein in the form of senile plaques and of tau protein in the form of neurofibrillary tangles. Amyloid β-protein (Aβ) is a 39–43 amino acid peptide cleaved from a larger precursor protein [amyloid precursor protein (APP)] found on chromosome 21.2 Aβ deposits extracellularly as senile plaques that can be visualized by haematoxylin and eosin staining (Figure 73.1b), with the fluorescent dye thioflavin S (Figure 73.1c), by immunohistochemistry using antibodies raised against Aβ epitopes (Figure 73.1d) or with silver impregnation (Figure 73.1e). Amyloid can appear either as loose, non-fibrillar diffuse plaques (Figure 73.1d) or a more compacted, fibrillar form (Figure 73.1b), often with dystrophic neurites coursing through the plaque (neuritic plaque, Figure 73.1e). Cerebral amyloid angiopathy (CAA) is due to the deposition of amyloid in the walls of arteries and arterioles and, less often, capillaries and veins of the central nervous system (Figure 73.1d). The walls of these vessels become very fragile and have a propensity to rupture, leading to superficial cortical haemorrhages. In addition to focal motor and sensory symptoms, repeated haemorrhages can lead to cognitive decline.
Figure 73.1 The neuropathology of AD. (a) Coronal sections of brain at the level of the hippocampus. On the left is a patient with AD, and on the right is an age-matched individual without cognitive impairment. Note the cortical atrophy and dilatation of the ventricles in the AD patient. (b) Extracellular Aβ senile plaques visualized with haematoxylin and eosin stain. (c) Thioflavin S fluorescent staining of amyloid plaques and neurofibrillary tangles. (d) Immunohistochemistry using Aβ antibodies demonstrating extracellular diffuse and fibrillar amyloid plaques and amyloid deposition in cerebral vessels. (e) Silver impregnation demonstrating fibrillar amyloid with dystrophic neurites and neurofibrillary tangles. (f) Immunohistochemistry using tau antibodies demonstrating neurofibrillary tangles and dystrophic neurites.
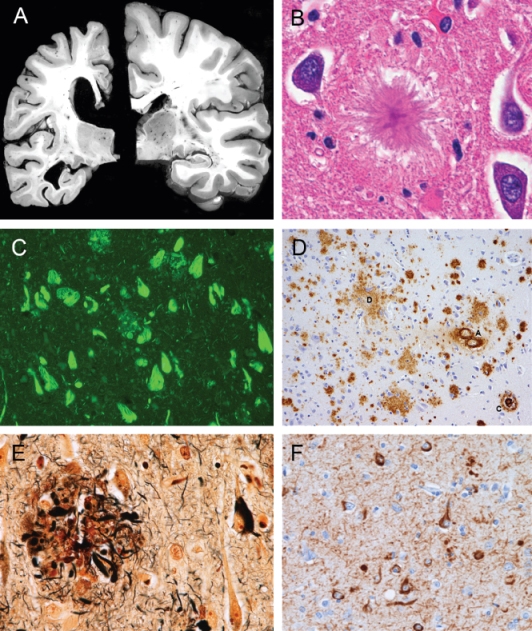
Tau protein is a microtubule-associated protein encoded on chromosome 17.2 Its normal function is to stabilize microtubules and it has numerous sites available for phosphorylation. When hyperphosphorylated, tau forms insoluble filaments that deposit in the cell body of the neuron as a neurofibrillary tangle (NFT) and in the axons and dendrites as neurophil threads (NTs). NFTs in AD are composed primarily of paired helical filaments (two strands of 10 nm diameter filaments that twist around each other like a helix). NFTs first appear in the hippocampus and entorhinal cortex in the AD brain (Figure 73.1f) and later involve the limbic and neocortex as the severity of dementia worsens. Hyperphosphorylated tau can be visualized by the same methods as amyloid since both proteins undergo transformations to a β-pleated sheet.
Epidemiology and Genetics
A number of risk factors have been associated with AD (Table 73.1).4 Age is clearly the most predictive risk factor. Although cases have been described as early as the third decade, the majority of cases occur after age 65 years. The prevalence of AD doubles each decade from 5% before age 65 years to nearly 50% at age 85 years. After age 85 years, studies are inconclusive as to whether the risk continues to increase; however, pathology characteristic of AD is frequently found in the brains of individuals over 90 years of age. In the oldest old, the course and progression of AD appear to be slower than for those individuals who develop the disease at younger ages. Family history of dementia in first-degree relatives appears to increase the risk of developing AD. Up to 25% of patients are able to identify a family member with the disease. There is an association between female gender and AD that persists after correction for differences in life expectancy between men and women. The reason for this remains unknown. Earlier epidemiological studies supported a role of postmenopausal estrogen levels; however, the Women’s Health Initiative found that estrogen replacement in later life increased rather than decreased the risk for AD.
Table 73.1 Alzheimer risk factors.
| Age |
| Female gender |
| Family history |
| Low education |
| Head injury |
| Apolipoprotein E ε4 allele |
| Cardiovascular risk factors: hypertension, diabetes, hyperlipidaemia, homocysteine |
| Late-life depression |
| Trisomy 21 (Down syndrome) |
| Mutations in amyloid precursor protein (APP), presenilin 1 (PS1) or presenilin 2 (PS2) |
Low educational attainment may also increase the risk of AD. There is increasing evidence supporting the hypothesis of cognitive reserve, that is, individuals with greater education are better able to stave off the effects of AD pathology in the early stages. Head injury associated with a loss of consciousness has been associated with increased risk of AD. Depression, particularly developing in late life, appears to be a prodromal symptom of AD. There is recent evidence to suggest that the same risk factors for cardiovascular disease may be important contributors to the risk of developing AD, including hypertension, high cholesterol, diabetes and homocysteine.1, 4 Epidemiological studies and chart-reviews have suggested that long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) may reduce the risk of AD; however controlled clinical trials have failed to replicate these results and instead suggest that the use of NSAIDs may increase risks of gastrointestinal, cardiovascular and cerebrovascular disease.
There are also several genetic risk factors. The best characterized risk for late-onset disease is associated with epsilon 4 (ε4) allele of apolipoprotein E (ApoE).5 ApoE is a cholesterol-carrying protein that may also play a role in handling of Aβ. Three isoforms of ApoE are present, ε2, ε3 and ε4. The ε3 allele is the most common, the ε2 is the least common and the ε4 allele is found in ∼20% of the population. The ε4 allele is over-represented in AD, with ∼60% of patients carrying at least one ε4 allele. Conversely, the presence of at least one ε2 allele appears to confer some protection. In Caucasians, compared with the ε3/ε3 genotype, individuals with one ε4 allele have a threefold increased risk whereas individuals with two ε4 alleles have a 15-fold increased risk of AD. This relationship holds true across most racial and ethnic groups.5 The linkage between ApoE and senile plaques suggests that cholesterol homeostasis may play either a direct or indirect role in the pathogenesis. However, because the ApoE genotype is not causative, genetic testing is not recommended as it neither improve diagnostic certainty nor changes the management of the disease. Additional risk genes are being investigated and validated with genome-wide association studies.6
Autosomal dominant, early-onset cases of AD are associated with mutations in the amyloid precursor protein (chromosome 21), presenilin 1 (chromosome 14) and presenilin 2 (chromosome 1).7 These three mutations appear to increase the production of the 42 amino acid long Aβ protein which has an increased propensity to aggregate. These mutations are rare, but can be suspected in a strong family history of AD with age of onset before 65 years. Even so, the mutations account for a small proportion of AD cases. The real value of the mutations has been in the creation of cell culture lines and transgenic animals that has greatly advanced research efforts. Associated with these genetic risk factors is the virtually certainty of AD developing in individuals with trisomy 21 (Down syndrome) with an additional copy of the region encoding the amyloid precursor protein. By the late 20s, Down syndrome patients begin to accumulate amyloid in their brains and nearly all patients are symptomatic by their mid-50s.1
Diagnostic Criteria
Although AD can only be definitively diagnosed by autopsy, current clinical criteria permit experienced clinicians to make accurate diagnoses most of the time: 92% or more of the time, an expert clinician’s diagnosis is confirmed by autopsy findings. The diagnosis of AD is one of inclusion using standardized clinical criteria: the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR)8 and the National Institute on Ageing—Alzheimer’s Association (NIA-AA).9 The DSM criteria are broader in nature and capture more impaired individuals whereas the NIA-AA criteria apply more rigorous definitions that incorporate new findings regarding the use of biological markers of disease. In general, the current diagnostic criteria are characterized by a two-step procedure with (1) the identification of a dementia syndrome and (2) the exclusion of other aetiologies of a dementia syndrome, using biological and neuroimaging examinations.
The diagnosis can also be based on the criteria of the DSM-IV-TR8 (Table 73.2). According to the DSM, the essential feature of dementia is impairment in short- and long-term memory, associated with impairment in abstract thinking, impaired judgment, other disturbances of higher cortical function or personality change. Disturbances in these cognitive domains should be severe enough to interfere with social or occupational functioning or disrupt interpersonal relationships with others. An important caveat in the DSM criteria is that the diagnosis cannot be made in the presence of a delirium.8 There are plans to update the DSM criteria but these are not available at the time of writing.
Table 73.2 DSM-IV criteria for dementia of the Alzheimer type.
| The development of multiple cognitive deficits manifested by both 1. Memory impairment (impaired ability to learn new information or to recall previously learned information) 2. One or more of the following cognitive disturbances: a. Aphasia (language disturbance) b. Apraxia (impaired ability to carry our motor activities despite intact motor function) c. Agnosia (failure to recognize or identify objects despite intact sensory function) d. Disturbances in executive functioning (i.e. planning, organizing, sequencing, abstracting) |
| The cognitive decline causes significant impairment in social or occupational functioning and presents a decline from a previous level of functioning |
| The course is characterized by gradual onset and continuing cognitive decline |
| The cognitive deficits above are not caused by any of the following: 1. Other central nervous system conditions that cause progressive deficits in memory and cognition 2. Systemic conditions that are known to cause dementia 3. Substance abuse conditions |
| The deficits do not occur exclusively during the course of a delirium |
| The deficits are not better accounted for by another Axis I disorder (depression, schizophrenia) |
Adapted from American Psychiatric Association, Task Force on DSM-IV. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV, 4th edn, American Psychiatric Association, Washington, DC, 1994.
According to NIA-AA guidelines, the diagnosis is first established by determining the presence of dementia. Dementia is defined as cognitive or behavioural symptoms that (1) interfere with social or occupational functioning, (2) represent a decline from previous level of functioning and (3) are not explained by another disorder. Dementia is established by a combination of a detailed history from the patient and a knowledgeable informant and an objective assessment of cognitive ability. Changes in cognitive ability should involve at least two of the following domains: memory, reasoning and problem solving, visuospatial abilities, language and/or changes in personality, behaviour or social comportment. Once the dementia syndrome is established, the diagnosis of probable AD can be made when the patient has (1) dementia, (2) an insidious onset with gradual progression and (3) worsening of symptoms over time either by observation or report of patient and/or informant. The presentation of deficits can either by amnestic (memory impairment) or non-amnestic (language, visuospatial, executive dysfunction).9 The diagnosis of AD should not be applied when there is clear evidence of cerebrovascular disease, Lewy body dementia, frontotemporal degeneration or another concurrent neurological, psychiatric or medical condition that can affect condition.9 If the patient meets criteria for probable AD but has evidence of other disorders that can affect cognition, a mixed dementia syndrome is most likely present.
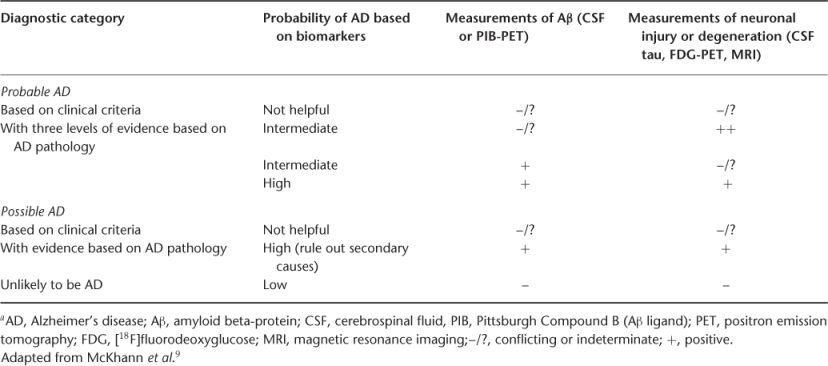
More importantly, the NIA-AA criteria take into consideration the role of biological markers of disease (biomarkers).10 Recent advances in biomarker research such as magnetic resonance imaging (MRI), positron emission tomography (PET) scans and cerebrospinal fluid (CSF) biomarkers characterize the underlying pathophysiological processes associated with AD such as evidence of Aβ deposition measured by CSF Aβ levels or PET scans or markers of neuronal injury by CSF tau levels, glucose hypometabolism by PET or cortical atrophy by MRI.9 Although the use of biomarkers is not advocated for routine clinical use, these tests may increase the diagnostic certainty of AD in difficult cases (Table 73.3).9
Table 73.3 Probability of AD with biomarkers (NIA-AA criteria)a.
Evaluation of the AD Patient
Clinical Evaluation
Knowing that the risk of AD increases with age, older adults are a natural choice for screening for AD. At present, however, there are no formal recommendations for or against dementia screening. The US Preventive Services Task Force (http://www.ahrq.gov/clinic/3rduspstf/dementia/dementrr.pdf) concluded that the evidence is insufficient to recommend for or against routine screening for dementia in older adults. Many of the current brief screening measures such as the Mini-Mental State Examination (MMSE)11 have good sensitivity but only fair specificity in detecting cognitive impairment and dementia.12, 13 The accuracy of the MMSE depends on a person’s age and educational level: using an arbitrary cut-point (typically ≤23) may potentially lead to more false-positives among older people with lower educational levels and more false negatives among younger people with higher educational levels.12
On the other hand, the early recognition of dementia, in addition to helping make diagnostic and treatment decisions, allows clinicians to anticipate problems the patients may have in understanding and adhering to recommended therapy. Early diagnosis is also beneficial to the patient’s caregiver(s) and family member(s) in helping to anticipate and plan for future problems that may develop as a result of progression of cognitive impairment and for long-term care. Long-term care planning and advanced care decisions are important in all forms of chronic diseases, many of which are treated by neurologists and psychiatrists. Discussions covering these topics early in the course of chronic disease should probably be considered part of the norm, rather than the exception. Organizations such as the American Medical Association, American Geriatrics Society, American Academy of Neurology and American Academy of Family Physicians all recommend that clinicians remain diligent in the early identification of symptoms of AD in their patients.
So who should be evaluated for AD? The Alzheimer Association has published 10 warning signs of symptoms that are most commonly seen in AD (Table 73.4).3 Although not every individual needs to be extensively worked up, developing a working list of reasons to consider AD as a possible diagnosis is reasonable (Table 73.5). Individuals with identified risk factors warrant further evaluation, as do individuals with memory complaints, with or without functional impairment. Additionally, even if an individual does not complain of memory or cognitive problems, informant complaints (spouses, adult children) should trigger further investigation. Evaluation should include a detailed history from the patient in addition to another source (spouse, caregiver, adult child) to gain insight into how the patient has changed from prior level of function. Historical points should highlight memory impairment (repetition; trouble remembering recent conversations, events, appointments; frequently misplacing items), executive function (deterioration of complex task performance; decreased ability to solve problems; difficulty with calculations; impaired driving), use of alcohol, prescription drugs and over-the-counter medications and the presence of focal neurological symptoms. A complete neurological examination will help to identify other causes of dementia (Table 73.6). Characteristic features of the four most common causes of dementia are given in Table 73.7 to help with differentiation.
Table 73.4 Ten warning signs.
| Memory loss |
| Difficulty in performing familiar tasks |
| Problems with language |
| Disorientation with respect to time and place |
| Poor or decreased judgement |
| Problems with abstract thought |
| Misplacing things |
| Changes in mood or behaviour |
| Changes in personality |
| Loss of initiative |
Source: Alzheimer’s Association.
Table 73.5 Indications for evaluating for AD.
Physician observations Difficulty in learning and retaining new information Difficulty in performing complex tasks Impaired reasoning Problems with orientation and spatial abilities Language difficulties, particularly word-finding Behaviour or personality changes Late-life depression, anxiety or apathy Previous well-controlled medical conditions now more difficult to manage Poor medication adherence/compliance |
Patient complaints Memory problems Work difficulties New-onset depression, anxiety or apathy Sleep changes (insomnia, nocturnal movements, unusual dreaming) |
Informant complaints Changes in memory or cognitive abilities Changes in functional abilities Changes inmood, personality or behaviour |
Table 73.6 Differential diagnosis of dementia.
Neurodegenerative disease Alzheimer’s disease Dementia with Lewy bodies/Parkinson’s disease dementia Frontotemporal dementia Huntington’s disease Progressive supranuclear palsy Corticobasal degeneration Multiple system atrophy Wilson’s disease Haemochromatosis/haemosiderosis Neuronal ceroid lipofuscinosis |