This article describes recent clinical and research advances in hemophilia therapy. Different prophylactic regimens for the management of severe hemophilia are described along with the use of adjuvant treatment options to achieve hemostasis. The safety and efficacy of radionuclide synovectomy with phosphorus 32-sulfur colloid to treat existing joint arthropathy also are described. The development of inhibitors to factor VIII or IX remains a challenge for hemophilia care and recent approaches to achieve immune tolerance induction are discussed. Finally, recent advances in hemophilia are mentioned, including the role of iron, inflammation, and angiogenesis in the pathogenesis of hemophilic arthopathy.
Hemophilia A or B is an X-linked recessive disorder that results from the deficiency of blood coagulation factor VIII or IX, respectively ( Fig. 1 ). Hemophilia is classified based on the level of factor VIII or IX activity as severe (<1%), moderate (1%–5%), or mild (>5%–40%). The type and frequency of bleeding in hemophilia vary according to its severity. For example, patients who have severe hemophilia present with spontaneous bleeding into the joints or muscles, soft tissue bleeding, and life-threatening hemorrhage in addition to episodes of minor bleeding. Patients who have moderate hemophilia present less commonly with spontaneous bleeding but frequently experience bleeding after minor trauma. Finally, patients who have mild hemophilia typically present bleeding only after surgery or major trauma.
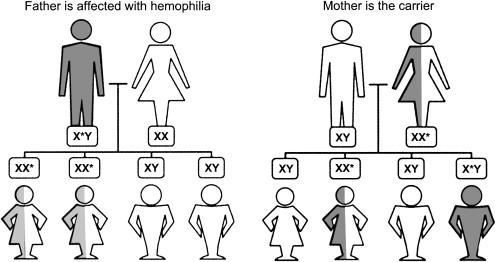
Musculoskeletal bleeding is the most common type of bleeding in hemophilia. Such bleeding can result in arthropathy, a common complication seen in the patient population that has this disease. In general, hemophilia treatment consists of replacing the missing coagulation protein with clotting factor concentrates when bleeding episodes occur (treatment on demand) or by scheduled infusions of clotting factor several times per week (prophylaxis). The development of neutralizing antibodies or inhibitors against factor VIII or IX is another complication encountered as a result of hemophilia treatment.
Multiple factor VIII and IX concentrates are available and categorized based on their source (plasma derived versus recombinant), purity, and viral inactivation methods. Recombinant products are categorized further based on the presence or absence of animal/human protein in the cell culture media or in the final stabilized product ( Table 1 ). Treatment of patients who develop inhibitors against factor VIII or IX is significantly more challenging than treatment of patients who do not have such antibodies. For patients who have high-titer inhibitors, the use of bypassing agents, such as recombinant factor VIIa or activated prothrombin complex concentrates, may be necessary to achieve hemostasis. In these particular patients, however, the ultimate therapeutic goal is to eradicate the inhibitor by means of immune tolerance induction (ITI). With this approach, patients receive repetitive doses of factor VIII or IX, usually once a day, with or without associated immunosuppresion. Typically, there is an initial rise in the antibody titers as a result of anamnestic response. Subsequently, however, a gradual reduction in titer is seen until in the end the inhibitor becomes undetectable. After successful immune tolerance, patients continue on regular factor infusions several times per week.
| Product Name (Manufacturer) | Viral Inactivation Procedures | Purity/Specific Activity (IU Factor VIII Activity/mg Total Protein) Before Addition of Stabilizer |
|---|---|---|
| Human plasma-derived factor VIII concentrates | ||
| Humate-P (ZLB Behring, Inc) (contains functional VWF protein) | Pasteurization (heating in solution, 60°C, 10 h) | Intermediate (1–10 IU/mg) |
| Alphanate SD (Grifols, Inc) (contains some functional VWF protein) |
| High (50–100 IU/mg) (>400 IU/mg after correcting for VWF content) |
| Koate-DVI (Bayer, Inc) (contains VWF protein) |
| High (50–100 IU/mg) |
| Monoclonal antibody-purified factor VIII concentrates (immunoaffinity purified from human plasma, no intact VWF protein) | ||
| Monarc-M (Baxter/Immuno, Inc using recovered plasma from the American Red Cross) |
| Ultra high (>3000 IU/mg) |
| Hemofil-M (Baxter/Immuno, Inc) |
| Ultra high (>3000 IU/mg) |
| Monoclate-P (ZLB Behring, Inc) |
| Ultra high (>3000 IU/mg) |
| Recombinant (genetic-engineered)/first-generation factor VIII concentrates | ||
| Recombinate (Baxter/Immuno, Inc) (human albumin as a stabilizer) |
| Ultra high (>4000 IU/mg) |
| Recombinant/second-generation factor VIII concentrates (human albumin-free final formulations) | ||
| Kogenate FS (Bayer, Inc) Helixate FS (Bayer for ZLB Behring, Inc) (sucrose as a stabilizer) |
| Ultra high (>4000 IU/mg) |
| Refacto (Wyeth, Inc) B-domain deleted (sucrose as a stabilizer) |
| Ultra high (>11,200–15,500 IU/mg), measured via chromogenic assay technique |
| Recombinant/third-generation factor VIII concentrates (no human or animal protein used in the culture medium or manufacturing process; does contain trace amounts of murine monoclonal antibody) | ||
| Advate (Baxter/Immuno, Inc) (trehalose as a stabilizer) |
| Ultra high (>4000–10,000 IU/mg) |
| Plasma-derived prothrombin complex concentrates/factor IX complex concentrates (nonactivated, also contain factor X and prothrombin but only traces of factor VII) | ||
| Bebulin VH (Baxter/Immuno, Inc) | Vapor heat (60°C, 10 h at 190 mbar pressure plus 1 h at 80°C, 375 mbar) | Intermediate (<50 IU/mg) |
| Profilnine SD (Grifols, Inc) | Solvent detergent (TNBP/polysorbate 80) | Intermediate (<50 IU/mg) |
| Proplex-T (Baxter/Immuno, Inc) | Dry heat (60°C, 144 h) | Intermediate (<50 IU/mg) |
| Plasma-derived prothrombin complex concentrates/factor IX complex concentrates (activated) | ||
| FEIBA (Baxter/Immuno, Inc) | Vapor heating (60°C, 10 h, 1160 mbar) | Intermediate (<50 U/mg) |
| Plasma-derived coagulation factor IX (human) concentrates | ||
| AlphaNine SD (Grifols, Inc) |
| High (>200 IU/mg) |
| Mononine (ZLB Behring, Inc) |
| High (>160 IU/mg) |
| Recombinant factor IX concentrates | ||
| BeneFIX (Wyeth, Inc) (no animal or human-derived protein in cell line; no albumin added to final product) |
| Ultra high (>200 IU/mg) |
| Factor VIII (or factor IX) concentrates useful in treatment of alloantibody and autoantibody inhibitor-related bleeding | ||
| Product name (manufacturer) | Viral inactivation method | Dosage |
| Recombinant factor VIIa (genetic engineered) | ||
| NovoSeven (Novo Nordisk, Inc) (stabilized in mannitol; bovine calf serum used in culture medium) |
| 90-μg/kg intravenous bolus every 2–3 h until bleeding ceases (larger dosing regimens are experimental but may be useful in refractory bleeding). This product is the treatment of choice for individuals who have allofactor IX antibody inhibitors and anaphylaxis or renal disease associated with the use of factor IX containing concentrates. |
| FEIBA-VH (Baxter Immuno, Inc) (human plasma derived) | Vapor heated (10 h, 60°C, 190 mbar plus 1 h, 80°C, 375 mbar) | 50–100 IU/kg not to exceed 200 IU/kg/24 h (for factor VIII and IX inhibitors) |
| Porcine plasma-derived factor VIII concentrate | ||
| Hyate C (Ibsen Biomeasure, Inc) | No longer available | >50 IU/mg (for factor VIII inhibitors only) |
This article focuses on recent advances. From a clinical standpoint, different prophylactic regimens, including primary, secondary, and tailored prophylaxis, for severe hemophilia are discussed. Some of these regimens may serve as alternatives to primary prophylaxis in developing countries where the high cost of factor concentrates precludes its regular use. Adjuvant treatment options for bleeding management in hemophilia also are discussed along with the role of radionuclide synovectomy with isotopes, such as phosphorus 32 sulfur colloid (P 32 ) to treat joint arthropathy. Current challenges in hemophilia care, including inhibitor development and approaches to achieve ITI, are addressed.
From a research standpoint, some of the mechanisms believed to lead to blood-induced joint disease are discussed. Data suggest that iron deposition in the synovium plays an important role in this process. This article discusses the experimental aspects of synovitis, including the role of iron and cytokines, in inducing an inflammatory response that stimulates angiogenesis and contributes to bone destruction.
Approaches to the medical management of hemophilia
Despite improvements in hemophilia therapy, arthropathy remains a significant clinical problem. The Medical and Scientific Advisory Council of the National Hemophilia Foundation, the World Federation of Hemophilia, and the World Health Organization all recommend that prophylaxis (intravenous factor replacement at least 46 weeks per year through adulthood infused in anticipation of and to prevent bleeding) be considered standard of care to prevent complications, such as arthropathy. Based on this recommendation, a major goal of hemophilia therapy is to prevent any joint disease.
Observational studies support that prophylaxis is superior to on-demand therapy in delaying or preventing the development of hemophilic arthropathy. There are different types of prophylaxis. Primary prophylaxis refers to therapy initiated in young patients who have hemophilia before joint damage (preventive therapy), whereas secondary prophylaxis refers to therapy initiated after joint abnormalities develop.
Since the 1990s, the standard of care for children who have severe hemophilia A or B in developed countries has been long-term prophylaxis, mainly primary, even though there is insufficient data to provide level A evidence of efficacy. Although experience over the past years shows the benefits of such approach, the high cost associated with primary prophylaxis has prevented it from being adopted in developing countries.
Manco-Johnson and colleagues recently published the results of the first prospective, randomized, controlled clinical trial in the United States evaluating the progression of arthropathy in children who have hemophilia treated with prophylaxis versus on-demand therapy. In this multicenter Joint Outcome Study, 65 children who had severe hemophilia (ages 30 months or less) were randomized to receive prophylaxis (25 IU/kg every other day) or an intense, episodic factor replacement (40 IU/kg initially, followed by 20 IU/kg at 24 and 72 hours after a joint bleed). Patients were followed until 6 years of age. Joint damage, the primary outcome of this study, was evaluated by MRI or plain radiographs. An 83% reduction in the risk for joint damage was shown by MRI in the prophylaxis group, supporting that such approach is effective in preventing joint damage. In 14% of cases of MRI changes, there was no evidence of any previous clinical hemarthrosis, suggesting that subclinical bleeding into the joints or the subchondral bone may cause joint damage. Further longitudinal imaging data likely are required to determine whether or not the use of continuous prophylaxis can prevent this subclinical bleeding from producing even minimal arthropathy years later.
Timing of Prophylaxis
The question of when to start primary prophylaxis has been a subject of controversy among hemophilia caregivers worldwide. To date, there is no consensus. Several studies show that children who have hemophilia and few or no joint bleeds who are started on prophylaxis early (mean age of 3 years) exhibit a better musculoskeletal outcome. Progression of joint arthropathy, even after starting prophylaxis, is described in patients who have at least five joint bleeds occurring at the same or different joints.
In a study published by Astermark and colleagues, the only significant predictor for development of hemophilic arthropathy was the age of patients when prophylaxis was started. Using the Pettersson score, a scoring system that allows radiologic evaluation of the joints in patients who have hemophilia, Fischer and colleagues described the Pettersson scores increasing by 8% each year that prophylaxis was postponed after the first occurrence of hemarthrosis. These studies show that irreversible joint damage may follow after a few joint bleeds and that even early prophylaxis may not abrogate that process completely. Therefore, worldwide recommendations to start prophylaxis before joint damage are favored to promote joint integrity, ideally before 3 years of age.
Secondary Prophylaxis
Patients who have preexisting joint disease and who experience frequent acute hemarthroses may be treated with periodic use of factor concentrates for a short or long period of time to curtail bleeding recurrence. This approach is known as secondary prophylaxis and is used commonly to minimize bleeding frequency and lessen the progression of joint disease. Even though secondary prophylaxis cannot reverse the changes of chronic arthropathy, it may be beneficial by reducing frequency of bleeding, hospital admissions, and lost days from school or work and by decreasing damage progression.
The use of secondary prophylaxis versus on-demand therapy has been the subject of various studies done in children and adults who have severe hemophilia. In summary, the results indicate that patients treated with secondary prophylaxis have decreased number of joint bleeds at the expense of higher clotting factor consumption. A recent study, however, does not confirm the higher cost of this approach.
A long-term outcome study (follow-up of 22 years) published by Fischer and colleagues compared the costs of prophylaxis (primary and secondary) versus secondary prophylaxis alone versus on-demand therapy in patients who had severe hemophilia. In this study, short-term prophylaxis was administered for 7.5 months (range 3 to 12 months) and long-term prophylaxis was administered over 12 months. Almost half of patients in the on-demand group occasionally had received short-term prophylaxis for several months to a year. This study showed that clotting factor consumption per year was similar for both treatment regimens (on-demand group: median of 1260 IU/kg per /year; prophylaxis group: median 1550 IU/kg per year). A significant difference, however, was that patients treated on demand presented a 3.2-fold increase in the frequency of joint bleeds, a 2.7-fold increase in clinical severity, and a 1.9-fold increase in Pettersson scores. Not surprisingly, the quality of life for this group of patients was decreased. Hence, these data support that the concept that prophylaxis may improve clinical outcomes without significantly increasing treatment costs.
Tailored Prophylaxis: Individualizing Therapy to Patients’ Needs
Because the natural history of arthropathy varies in patients who have hemophilia, even considering those who are classified as having severe hemophilia, tailoring prophylaxis to a patient’s bleeding pattern, joint involvement, and individual needs seems a reasonable approach.
As discussed previously, data support initiating prophylaxis at an early age to prevent joint damage. Particularly in young children, however, establishing venous access commonly is a challenge and not surprisingly central venous catheters (CVCs) often are required in this population for the administration of multiple dosages of factor. The benefit of easy access provided by such catheters must be balanced by their risks, in particular catheter-related infection.
Several studies have described different regimens to initiate prophylaxis early on with a dual goal of preventing joint damage and minimizing the need for CVC placement in young children. For example, Petrini reported that primary prophylaxis can be started using a weekly infusion of factor concentrate rather than the standard 3-times-per-week prophylactic regimen as early as 1 or 2 years of age. This approach reduces the need for CVC placement in young children without increasing the occurrence of hemarthrosis. Astermark and colleagues reported in a similar study that there was no difference in the occurrence of hemarthrosis or arthropathy when comparing children who received factor VIII concentrate infusions weekly during their first year of prophylaxis versus those who received prophylaxis 3 times a week.
In another study, published by van den Berg and colleagues, outcomes of tailored prophylaxis were described for three cohorts according to the time at which prophylaxis was started in relationship to the number of joint bleeds. Data from this study also support the use of tailored prophylaxis to prevent hemophilic arthropathy after a first joint bleed has occurred.
A prospective, multicenter Canadian study enrolling children who have hemophilia is ongoing to evaluate frequency of infusions and dose escalation. In this study, 25 children who had severe hemophilia A (ages 1 to 2.5 years) and normal joints were enrolled on a dose-escalation protocol defined by breakthrough hemarthroses. With this regimen, the dose of factor VIII concentrate increased from a weekly dose of 50 IU/kg to a twice-a week-dose of 30 IU/kg to an every-other-day dose of 25 IU/kg determined by the frequency of breakthrough musculoskeletal bleeds. An interim report of this cohort has been described after a median follow-up of 4.1 years. Of the 25 patients enrolled, 13 (52%) required a dose escalation to twice-a-week prophylaxis secondary to frequent joint bleeds (median time to escalation 3.42 years). Alternatively, dose escalation had not been required in 12 (48%) of these patients. The occurrence of a target joint, however, one in which recurrent bleeding has occurred on at least four occasions during the previous 6 months or where 20 lifetime bleeding episodes have occurred, was evident in 40% of the patients. This indicates that the long-term effect on joint outcome using this approach warrants further scrutiny.
Adjuvant treatment options for patients who have hemophilia
Desmopressin (DDAVP) has been used to control or prevent bleeding in mild hemophilia A, some cases of moderate hemophilia A, and some types of von Willebrand disease since 1977. Its mechanism of action seems multifactorial, including an increase in plasma levels of factor VIII and von Willebrand factor (VWF), stimulation of platelet adhesion, and increased expression of tissue factor. DDAVP is not effective for the treatment of patients who have hemophilia B, as factor IX levels are not influenced by DDAVP.
For mild to moderate hemophilia and von Willebrand disease, the indications for DDAVP use are determined by the type of bleeding episode, baseline, and desired level of factor VIII and VWF. A test dose, also known as DDAVP challenge, should be performed under controlled conditions, such as in a doctor’s office, where blood pressure and heart rate can be monitored. A blood sample is obtained before the test dose and approximately 2 hours after administration of the test dose. A twofold to fourfold rise in the levels of factor VIII, von Willebrand antigen, and ristocetin cofactor activity is expected. This expected rise in factor VIII levels explains why patients who have severe hemophilia A are not candidates for this type of therapy to control bleeding. The intranasal route is the administration route of choice for outpatient treatment and commonly is used before dental procedures and oral/nasal mucosal bleeding. The intravenous route also is available and typically used in the inpatient setting. One advantage of intravenous administration is that peak levels tend to be higher and achieved faster. Patients should be advised to limit water intake during DDAVP treatment and to avoid using more than three consecutive daily doses to reduce the risk for developing hyponatremia.
Antifibrinolytic agents, epsilon-aminocaproic acid and tranexamic acid, both lysine derivatives, also are useful adjuvant therapy for patients who have mild to severe hemophilia. They exert their effect by inhibiting the proteolytic activity of plasmin and, therefore, inhibiting fibrinolysis. The use of antifibrinolytic agents is indicated in the presence of mucosal bleeding, primarily oral, nasal, and menstrual blood loss. Its use is contraindicated in presence of hematuria because of increased risk for intrarenal or ureteral thrombosis, in the presence of disseminated intravascular coagulation, or thromboembolic disease. Both drugs are available for use in the United States in an intravenous form (epsilon-aminocaproic acid at 100 mg/kg/dose every 6 hours; tranexamic acid at 10 mg/kg/dose every 6–8 hours). Currently, only epsilon-aminocaproic acid is available for use in the United States in an oral form.
Topical agents, such as fibrin sealant, which is prepared by mixing two plasma-derived protein fractions (fibrinogen-rich concentrate and thrombin concentrate), are used for local bleeding control in hemophilia. There are concerns, however, regarding the use of bovine thrombin in this setting. First, there seems to be a high rate (approximately 20% or higher) of antibody formation against thrombin and factor V, which may inactivate an individual’s own endogenous factor V or thrombin production, thereby creating a new bleeding diathesis. Another legitimate concern is the potential risk for transmitting blood-borne pathogens, such as variant Creutzfeldt-Jakob virus. When available commercially, the use of recombinant human thrombin will likely minimize these risks.
A phase 3, prospective, randomized, double-blind, comparative study evaluating the safety and efficacy of topical recombinant human thrombin and bovine thrombin in surgical hemostasis has been published. The primary objective of this study was to evaluate the efficacy of both products whereas the secondary objective was to evaluate safety and antigenicity. This multicenter study enrolled more than 400 patients, who were randomized in a 1:1 ratio. The study showed that both topical agents had a comparable efficacy of 95% with similar adverse events rates. A statistically significant lower incidence of antibodies, however, was identified against recombinant human thrombin (1.5%) compared with antibovine thrombin (21.5%). Based on the results of this study, recombinant human thrombin seems the preferred option to achieve topical hemostasis.
Recombinant factor VIIa is a Food and Drug Administration–approved product used to promote hemostasis in patients who have hemophilia A or B with inhibitors. Its mechanism of action includes binding of activated factor VII to tissue factor, which activates factor X and leads to thrombin generation. Recombinant factor VIIa also binds the surface of activated platelets independent of tissue factor, activating factor X and leading to thrombin generation. The standard dose in hemophilia with inhibitors is 90 to 120 μg/kg every 2 to 3 hours until hemostasis is achieved. Subsequent dosing and interval is based on clinical judgment. At the present time, there is no validated laboratory test to monitor recombinant factor VIIa therapy. Clinical experience shows an excellent or effective response in more than 90% of patients who have hemophilia and low risk for thrombosis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


