div class=”ChapterContextInformation”>
6. The Adrenal Glands
6.1 Adrenal Glands: Anatomy and Physiology of Adrenal Cortex
a medulla → epinephrine (E), norepinephrine (NE), small amounts of dopamine
a cortex → the adult cortex is composed of three zones: an outer zona glomerulosa, a zona fasciculata, and an inner zona reticularis
Zones and Steroidogenesis
Because of the enzymatic differences between the zona glomerulosa and the inner two zones, the adrenal cortex functions as two separate units, with differing regulation and secretory products. Thus, the zona glomerulosa, which produces aldosterone, lacks 17α-hydroxylase activity and cannot synthesize 17α-hydroxypregnenolone and 17α-hydroxyprogesterone, which are the precursors of cortisol and the adrenal androgens.
The zona fasciculate and zona reticularis produce cortisol, androgens, and small amounts of estrogens. These zones, primarily regulated by ACTH, do not express the gene CYP11B2 (encoding P450aldo) and therefore cannot convert 11-Deoxycorticosterone (DOC) to aldosterone.
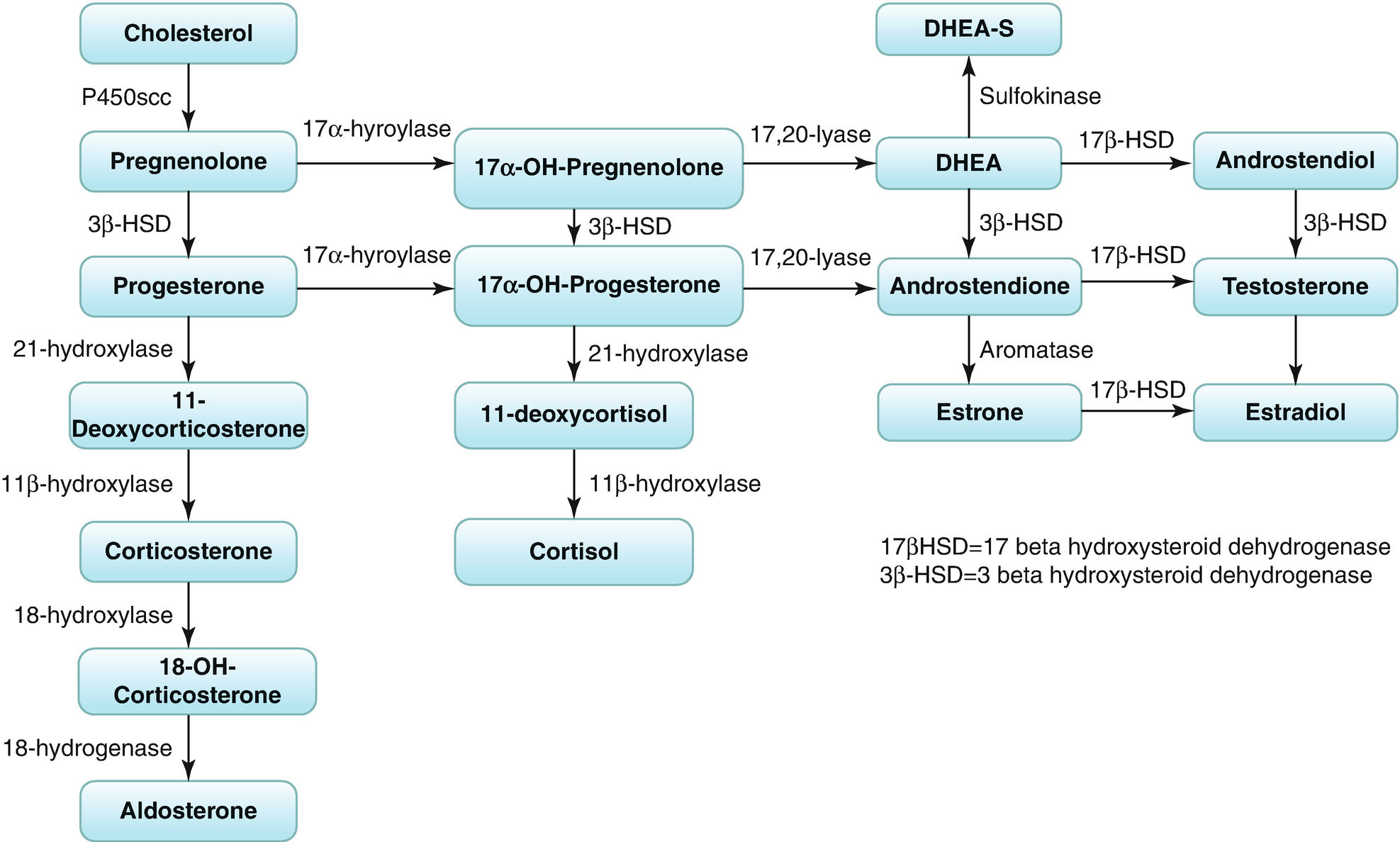
Biosynthesis of corticosteroids and sex steroids
6.2 Glucocorticoids, Adrenal Androgens, and Aldosterone: Regulation of Secretion
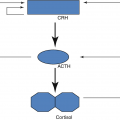
Secretion of CRH and ACTH
ACTH is the trophic hormone of the zona fasciculate and reticularis and the major regulator of cortisol and adrenal androgen production; excess or deficiency of this hormone alters their structure and function. Thus, both zones atrophy when ACTH is deficient; when ACTH is present in excess, hyperplasia and hypertrophy of these zones occur.
ACTH, in turn, is regulated by the hypothalamus and central nervous system via neurotransmitters and CRH and arginine vasopressin.
Neuroendocrine Control
- 1.
Episodic secretion and the circadian rhythm of ACTH
Circadian rhythm is superimposed on episodic secretion; it is the result of central nervous system events that regulate both the number and magnitude of CRH and ACTH secretory episodes. Cortisol secretion is low in the late evening and continues to decline, so around 12.00–1.00 a.m. cortisol levels may be undetectable. During the third and fifth hours of sleep there is an increase in secretion; but the major secretory episodes begin in the sixth to eighth hours of sleep and then begin to decline as wakefulness occurs. About half of the total daily cortisol output is secreted during this period. Cortisol secretion then gradually declines during the day, so values obtained later in the day are lower and at 4 p.m. are approximately half of morning values.
- 2.
Stress responsiveness of the hypothalamic–pituitary–adrenal (HPA) axis
Responses to stress (e.g., surgery and hypoglycemia) originate in the central nervous system and increase hypothalamic CRH and thus pituitary ACTH secretion.
- 3.
Feedback inhibition by cortisol of ACTH secretion
The third major regulator of ACTH and cortisol secretion is that of feedback inhibition by glucocorticoids of CRH, ACTH, and cortisol secretion.
Adrenal Androgens
Adrenal androgen production in adults is also regulated by ACTH; on the contrary, the androgens (both DHEA and androstenedione) do not regulate through feedback inhibition by the ACTH secretion. The direct biologic activity of the adrenal androgens (androstenedione, DHEA, and DHEA sulfate) is minimal, and they function primarily as precursors for peripheral conversion to the active androgenic hormones, testosterone, and dihydrotestosterone. Thus, DHEA sulfate secreted by the adrenal androgens undergoes limited conversion to DHEA; this peripherally converted DHEA and that secreted by the adrenal cortex can be further converted in peripheral tissues to androstenedione, the immediate precursor of the active androgens.
Aldosterone
The synthesis of aldosterone by the zona glomerulosa is primarily regulated by the renin–angiotensin system and by potassium (+ to a lesser extent by ACTH as well).
Adrenocortical Insufficiency
Deficient adrenal production of glucocorticoids or mineralocorticoids results in adrenocortical insufficiency, which is either the consequence of destruction or dysfunction of the cortex (primary adrenocortical insufficiency (PAI), or Addison disease and congenital adrenal hyperplasia (CAH)) or secondary to deficient pituitary ACTH secretion (secondary adrenocortical insufficiency, discussed elsewhere, see section hypopituitarism). Glucocorticoid therapy is the most common cause of secondary adrenocortical insufficiency.
6.3 Primary Adrenocortical Insufficiency (Addison Disease)
6.3.1 Definition
Adrenocortical insufficiency (ACI) is a rare disease having a prevalence of 10–15 per 100,000 population, characterized by deficient adrenal production of glucocorticoids or mineralocorticoids.
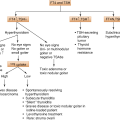
6.3.2 Etiology of Primary ACI (Table 6.1)
Etiology (excluding CAH) and pathology of primary ACI
Autoimmune (the most common cause in developed countries—70%) • Sporadic • Type 1 or 2 autoimmune polyendocrine syndromes (APS) |
Infiltrations: Amyloidosis, hemochromatosis, sarcoidosis |
Infections: Tuberculosis (prior to 1920, was the major cause), funguses, CMV, HIV |
Adrenal hemorrhage/infarction: • Anticoagulant therapy • Waterhouse–Friderichsen syndrome (meningococcal septicemia) • The primary antiphospholipid antibody syndrome (lupus anticoagulant) |
Congenital adrenal hypoplasia (mutations/deletion of the NR0B1(DAX-1) or the NR5A1 (SF-1) genes) |
Adrenoleukodystrophy (X-linked, is an important cause of ACI in men; abnormal accumulation of very long chain fatty acids (VLCFAs) in the brain, adrenal cortex, testes, and liver) |
Metastatic adrenal disease (common sites of metastasis for lung, gastrointestinal, breast, and renal neoplasia) |
Iatrogenic • Bilateral adrenalectomy • Drugs: ketoconazole, abiraterone, etomidate and aminoglutethimide (inhibit cortisol biosynthesis), mitotane (adrenolytic) |
APS, Type I
Autosomal recessive with childhood onset.
Mutations in the AIRE (autoimmune regulator) gene located on chromosome 21p22.3.
Hypoparathyroidism, chronic mucocutaneous candidiasis, ACI, gonadal failure, hypothyroidism, type 1 DM, DI+ nonendocrine diseases: alopecia, pernicious anemia, chronic active hepatitis.
Autoantibodies against the cholesterol side chain cleavage enzyme (P450scc, CYP11A1) and others have been described.
APS Type II
Adult onset.
ACI, autoimmune thyroid disease, gonadal failure, type 1 DM, DI+ celiac sprue, vitiligo, pernicious anemia, myasthenia gravis (also known as Schmidt’s syndrome).
Polygenic inheritance, association with HLADR3 and CTL4 regions, mixed penetrance.
These patients have antiadrenal cytoplasmic antibodies and autoantibodies directed against 21-hydroxylase (P450c21, CYP21A2).
6.3.3 Clinical Diagnosis of Primary ACI (Addison Disease)
- 1.
Hyperpigmentation is the classic physical finding. Generalized hyperpigmentation of the skin and mucous membranes is one of the earliest manifestations of Addison disease. It is increased in sun-exposed areas and accentuated over pressure areas such as the knuckles, toes, elbows, and knees. The classic hyperpigmentation of the buccal mucosa and gums is preceded by generalized hyperpigmentation of the skin; ACI should also be suspected when there is increased pigmentation of the palmar creases, nail beds, nipples, areolae, and perivaginal and perianal mucosa. Scars that have formed after the onset of ACTH excess become hyperpigmented, whereas older ones do not.
- 2.
General weakness and fatigue, malaise, anorexia, weight loss (!!! 15 kg) are invariable features of the disorder.
- 3.
Cardiovascular symptoms: Hypotension is present in about 90% of patients, accompanied by orthostatic symptoms: dizziness and occasionally syncope.
Positive Schellong test (a reduction in systolic BP of at least 20 mmHg or diastolic BP of at least 10 mmHg within 3 min of standing).
- 4.
Gastrointestinal disturbances: Nausea, vomiting, diarrhea or constipation, abdominal pain.
- 5.
Others:
- (a)
Salt craving occurs in about 20% of patients.
- (b)
Arthralgia; myalgia.
- (c)
Hypoglycemia (→ sweats, tremor, headache) more commonly in secondary adrenal insufficiency and children.
- (d)
Irritability, emotional instability, depression.
- (e)
Loss of axillary and pubic hair-in ♀ (←↓ secretion of adrenal androgens).
- (f)
Vitiligo occurs in 4–17% of patients with autoimmune Addison disease.
- (a)
Clinical Features in Secondary Adrenal Insufficiency
Hyperpigmentation is not present—on the contrary, the skin is pale.
Mineralocorticoid secretion is usually normal.
6.3.4 Paraclinical Diagnosis of Primary Adrenocortical Insufficiency
The diagnosis of PAI requires two steps.
6.3.4.1 First Step
Normal value cortisol
8 a.m. 5–25 μg/dL (171–536 nmol/L)
6–8 p.m. 2.5–13.0 μg/dL (71.1–286 nmol/L)
12 a.m. <7 μg/dL
The diagnostic test for primary adrenocortical insufficiency should be paired measurement of serum cortisol and plasma ACTH.
Undetectable serum cortisol is diagnostic of adrenal insufficiency, primary or secondary (and undetectable cortisol + elevated ACTH indicates PAI), while a cortisol >550 nmol/L precludes the diagnosis.
However, in many patients, simultaneous 9 a.m. cortisol and ACTH will show an elevated ACTH for the level of cortisol, which is often in the normal range or low-normal. This is a very sensitive means of detecting Addison’s disease.
In equivocal cases (e.g., when uncertainty exists whether or not partial primary ACI is present), a cosyntropin (synacthen or tetracosactide) test may be necessary.
The rapid ACTH stimulation test (synthetic human 1–24-ACTH called tetracosactrin or cosyntropin is used) measures the acute adrenal response to ACTH and is used to diagnose both primary and secondary adrenal insufficiency.
→ A Synacthen stimulated (0.25 mg IM or IV) peak serum cortisol <500 nmol/L is diagnostic of PAI (Note: a similar response can be observed in overt secondary adrenal insufficiency).
Normal responses (peak cortisol, 30–60 min later, >550 nmol/L (20 μg/dL)):
A normal response to the rapid ACTH stimulation test excludes both primary adrenal insufficiency (by directly assessing adrenal reserve) and overt secondary adrenal insufficiency with adrenal atrophy.
However, a normal response does not rule out partial ACTH deficiency (decreased pituitary reserve) in patients whose basal ACTH secretion is sufficient to prevent adrenocortical atrophy and patients with recently developed secondary adrenal insufficiency who have not yet undergone adrenal atrophy.
These patients may be unable to further increase ACTH secretion and thus may have subnormal pituitary ACTH responsiveness to stress or hypoglycemia. In such patients, further testing with hypoglycemia (as already discussed in the hypothalamus–pituitary section) may be indicated.
Plasma ACTH 8–9 a.m.
In adrenal insufficiency due to primary adrenal disease, plasma ACTH levels are elevated.
Conversely, in pituitary ACTH deficiency and secondary hypoadrenalism, plasma ACTH levels are inappropriately normal or less than 10 pg/mL (2.2 pmol/l).
Additional Laboratory Investigations
Hyponatremia and hyperkalemia are classic manifestations of glucocorticoid and mineralocorticoid deficiency and should suggest the diagnosis—K ↑↑ (acute adrenal crisis).
FBC—normocytic, normochromic anemia (← altered androgen metabolism) neutropenia, eosinophilia, and a relative lymphocytosis.
↑ Concentration of blood urea nitrogen and serum creatinine (← volume depletion and dehydration).
Mild acidosis, mild hypercalcemia.
Serum glucose ↓<70 mg/dL.
ECG: low voltage, a vertical QRS axis, and nonspecific ST-T wave abnormalities secondary to abnormal electrolytes.
6.3.4.2 Second Step
Once PAI is confirmed, the etiology should be established.
Tests for Etiology
Adrenal autoantibodies (detect antibodies to adrenal cortex and, specific antibodies to 21-hydroxylase, side chain cleavage enzyme, and 17-hydroxylase) (indirect information ← testing for thyroid autoantibodies).
Imaging:
Adrenal enlargement, with or without calcification, may be seen on CT of the abdomen, suggesting tuberculosis, infiltration, or metastatic disease.
The adrenals are small and atrophic in chronic autoimmune adrenalitis.
Percutaneous CT-guided adrenal biopsy is occasionally required (→ metastatic lesions suspected).
Specific tests, for example, serological or microbiological investigations directed at particular infections (e.g., tuberculosis)/VLCFAs (adrenoleukodystrophy).
6.3.5 Treatment of Chronic ACI
6.3.5.1 Objectives
Long-term: to produce levels of glucocorticoids and mineralocorticoids equivalent to those achieved in an individual with normal HPA function under similar circumstances.
Patient education–education on how to increase steroid doses during concurrent illnesses or injury is important.
6.3.5.2 Maintenance Therapy
General principles: Patients with PAI require lifelong steroid replacement, both glucocorticoids and mineralocorticoids, and often an increased intake of sodium chloride to compensate for increased renal loss.
Glucocorticoid Replacement
Glucocorticoids are secreted into the systemic circulation in a pulsatile (ultradian) and circadian fashion, with a peak in the morning and reaching a nadir at midnight. Individuals with normal adrenal function produce between 5 and 10 mg of cortisol per m2 of body surface area per day, equivalent to an oral replacement dose of 15–25 mg/day of hydrocortisone.
Hydrocortisone (HC) or cortisone acetate (CA) is the treatment of choice for replacement therapy, administered twice to thrice daily, for example, 10 mgHC immediately on waking, 5 mgHC at midday, and 5 mgHC at 4–5 p.m., or 15 mgHC on waking and 5 mgHC at midday.
Alternative: Prednisone 5–7.5 mg/day.
Note: A modified release hydrocortisone formulation (Plenadren) allowing once daily dosing has recently been introduced in Europe.
Mineralocorticoid Replacement
9-α-Fluorocortisol (fludrocortisone) (Astonin H) 0.1 mg/tb, 0.05–0.2 mg/day is used for mineralocorticoid therapy; ! 10% can be managed with cortisol and dietary sodium intake alone; rarely required in central adrenocortical insufficiency.
40 mg hydrocortisone has the equivalent mineralocorticoid effect of 100 μg fludrocortisone.
DHEA Replacement
DHEA replacement (25–50 mg/day) may improve mood and well-being as well as libido in women with ACI.
Lifestyle advice—↑ in proteins, vitamin, adequate dietary sodium intake (!!! not restriction).
6.3.5.3 Monitoring of Therapy
Clinical
Adequate treatment results in the disappearance of weakness, malaise, and fatigue, anorexia and other gastrointestinal symptoms; the hyperpigmentation invariably improves.
Inadequate cortisol administration leads to persistence of these symptoms of adrenal insufficiency, and excessive pigmentation remains/conversely, overtreatment → signs of Cushing syndrome, for example, ↑ weight.
BP (including postural change).
Hypertension and edema suggest excessive mineralocorticoid replacement whereas postural hypotension and salt craving suggest insufficient treatment.
Biochemical
Serum electrolytes.
Plasma renin (elevated if insufficient fludrocortisone replacement).
Cortisol day curves + ACTH, or plasma cortisol to assess adequacy have been proposed but not yet widely adopted.
6.3.5.4 General Measures: Prevention of the Adrenal Crisis
The essential elements are patient education and ↑ glucocorticoid dosage during illness.
During period of minor stress → the daily dose of HC should be doubled (or 3×).
The dose needs to be ↑ to as much as 200–400 mg HC/day during periods of major stress, such as a surgical procedure.
6.4 Acute Adrenocortical Insufficiency (Addison Crisis)
6.4.1 Definition
Acute adrenal crisis represents a state of acute adrenocortical insufficiency and occurs in patients with Addison disease who are exposed to the stress of infection, trauma, surgery, or dehydration due to salt deprivation, vomiting, or diarrhea. Bilateral acute adrenal hemorrhage and CAH represent another two possible causes.
6.4.2 Clinical Picture
Hypotension and shock, volume depletion, dehydration, hyperpyrexia (← infection or hypoadrenalism per se), weakness, apathy, depressed mentation, abdominal pain (that mimic an acute abdominal emergency) (abdominal distention, rigidity, and rebound tenderness are less frequent), nausea, vomiting, anorexia, muscle cramps, hypoglycemia. Shock and coma may rapidly lead to death in untreated patients.
6.4.3 Laboratory Findings
Additional findings that suggest the diagnosis are hyponatremia, hyperkalemia, lymphocytosis, eosinophilia, and hypoglycemia, urea ↑, creatinine ↑, hematocrit ↑, metabolic acidosis + ↓↓ CORTISOL.
6.4.4 Management
Emergency!
Take blood for glycemia, electrolytes, urea, creatinine, cortisol before initiating treatment with glucocorticoids.
IV 0.9% saline and 5% glucose are administered to correct volume depletion, hypotension and hypoglycemia.
Hydrocortisone should be administered in a dosage of 100 mg intravenously followed by 50–75 mg every 6 h thereafter; when the patient is stable, reduce the dosage to 50 mg every 6 h; mineralocorticoid is not necessary during the acute replacement period since enough NaCl and glucocorticoid are being administered to treat the mineralocorticoid deficiency; taper to maintenance therapy by day 4 or 5 and add mineralocorticoid.
Identify and treat precipitating factor (e.g., infection).
6.5 Congenital Adrenal Hyperplasia
Definition
>90% of cases are due to 21α-hydroxylase deficiency.
6.5.1 P450c21 Hydroxylase Deficiency
6.5.1.1 Presentation
Carrier frequency of classic CAH 1:60–1:100 in Caucasians.
Carrier frequency of nonclassic CAH 19% in Ashkenazi Jews, 13.5% in Hispanics, 6% in Italians, and 3% in other Caucasian populations.
Clinical picture is a consequence of the degree of enzymatic deficiency and ranges from presentation in neonatal period with salt wasting and virilization to nonclassic CAH in adulthood and depends on severity of mutation.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree