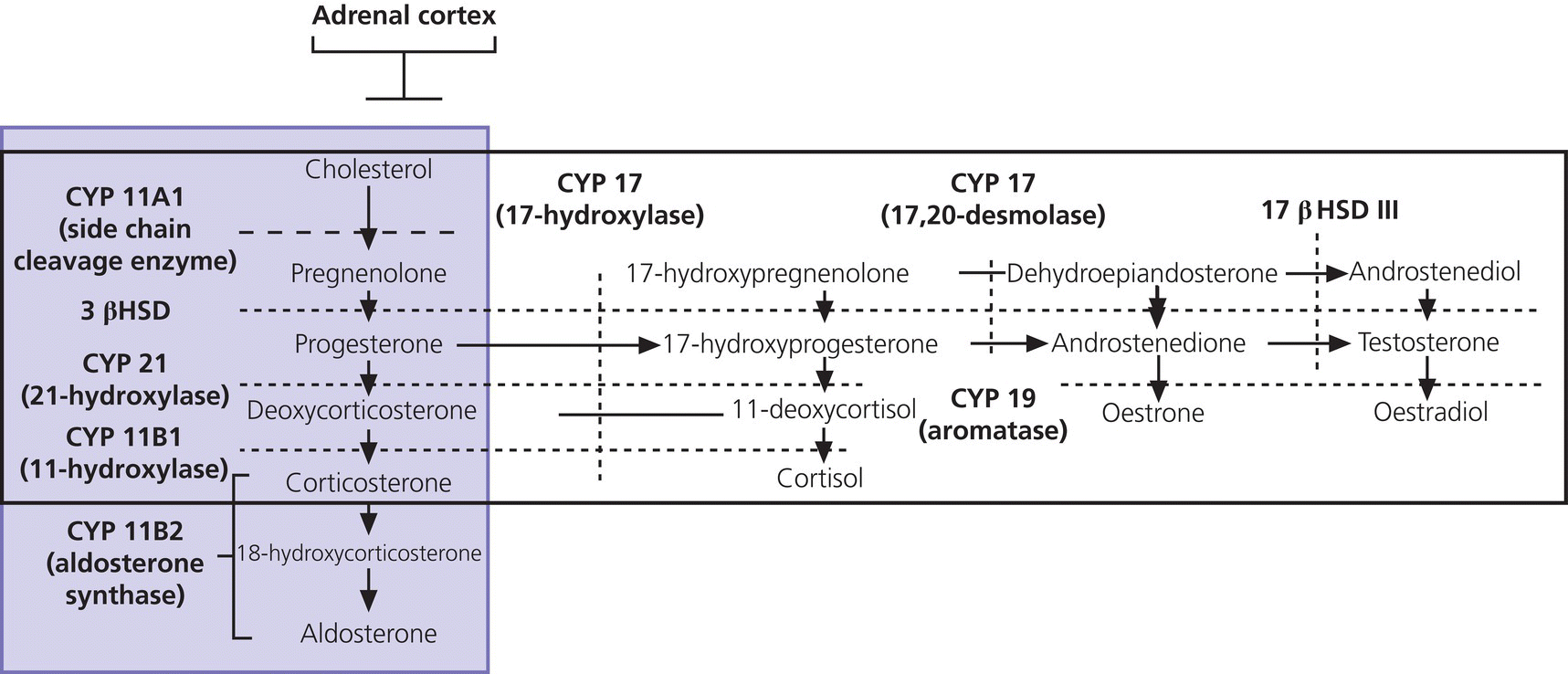
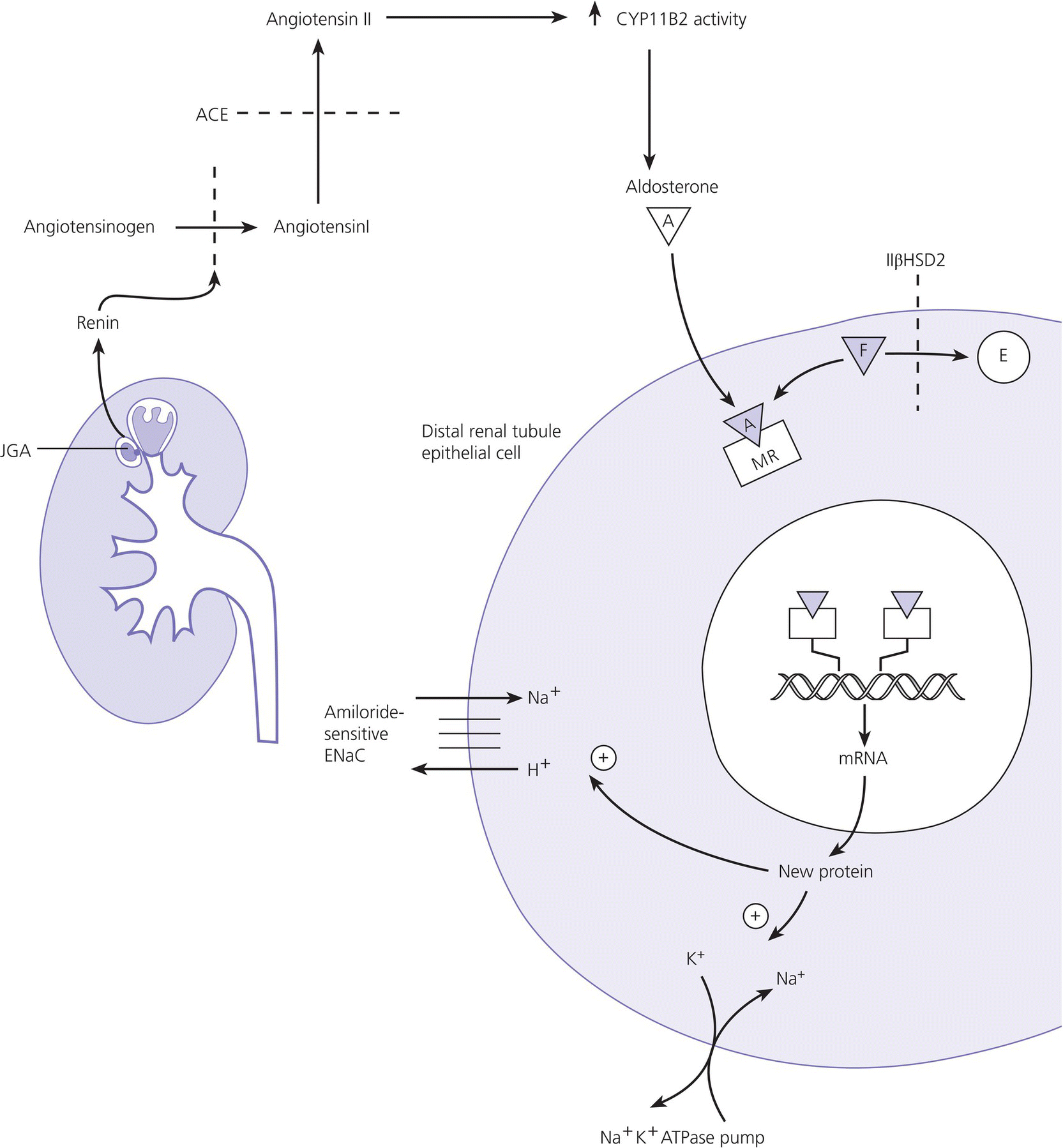
8 The adrenal glands are pyramidal structures which lie adjacent to the upper poles of the kidneys. The adrenal cortex is divided into three zones: the outer zona glomerulosa, the middle zona fasciculata, and the inner zona reticularis. The main types of steroids produced by the adrenal cortex are mineralocorticoids (principally aldosterone), glucocorticoids (principally cortisol), and androgens. Steroidogenesis in the zona fasciculata (primarily cortisol) and reticularis (mainly androgen) is under hypothalamic–pituitary control (Figure 8.1); secretion of aldosterone from the zona glomerulosa is under the control of the renin–angiotensin system. Figure 8.1 Schematic diagram of hypothalamic–pituitary–adrenal axis. b LPH, b lipotrophic hormone; CRH, corticotrophin‐releasing hormone; JP, joining peptide; N‐T peptide, N‐terminal peptide; POMC, pro‐opiomelanocortin; StAR protein, steroidogenic acute regulatory protein; +, positive effect; −, negative effect. Corticotrophin‐releasing hormone (CRH) is synthesized in the hypothalamus and acts on the corticotrophs of the anterior pituitary to secrete the peptide proopiomelanocortin (POMC). POMC is the precursor of adrenocorticotrophic hormone (ACTH). ACTH binds to its melanocortin‐2 receptor (MC‐2R), a cell surface receptor in the cells of the adrenal cortex. ACTH also has affinity for the melanocortin‐1 receptor (MC‐1R) in the skin, so that ACTH excess results in hyperpigmentation. The MC‐2R is G‐protein coupled, and its activation results in increased adenylate cyclase activity, which in turn increases intracellular cyclic adenosine monophosphate (cAMP). Increased cAMP activity enhances the transport of cholesterol across the mitochondrial membrane by the steroidogenic acute regulatory (StAR) protein. Cholesterol is metabolized into three types of steroids: (i) glucocorticoid; (ii) mineralocorticoid; and (iii) androgen. Cortisol, the principal glucocorticoid, is vital for maintaining normal glucose and blood pressure and for combating stress. It follows a circadian rhythm, with high levels on waking and a nadir at midnight. Between 80% and 90% of cortisol is bound to cortisol binding globulin (CBG), and most plasma assays measure total rather than free cortisol. Cortisol exerts a negative feedback at the hypothalamic and pituitary levels. Thus, plasma ACTH concentrations will be elevated in primary adrenal insufficiency and suppressed when cortisol excess is caused by autonomous adrenal lesions or exogenous glucocorticoids. In primary adrenal insufficiency, cortisol levels will depend on the severity of the defect. In severe deficiency, cortisol is low despite massively raised ACTH, but in partial deficiency, basal cortisol may be normal. However, since basal cortisol output is already maximal in this situation, there will be no further increase should a major physical stress occur and signs of acute adrenal insufficiency may then develop. Adrenal steroidogenesis is under the control of the cytochrome P450 (CYP) and the hydroxysteroid dehydrogenase (HSD) enzymes. Pregnenolone is converted along the mineralocorticoid pathway to aldosterone, along the glucocorticoid pathway to cortisol, or along the androgen pathway to testosterone (Figure 8.2). Impairment of cortisol synthesis results from disorders affecting the StAR protein and 3‐β‐hydroxysteroid dehydrogenase (3‐βHSD), CYP 21A2 (21‐hydroxylase), CYP 11B1 (11‐hydroxylase), and the CYP 17 (17‐hydroxylase) enzymes. These enzyme disorders are collectively known as ‘congenital adrenal hyperplasia (CAH)’, as cortisol deficiency causes increased ACTH secretion and thus enlargement of the adrenal glands. Mineralocorticoid deficiency results from StAR protein, 3‐βHSD and CYP 21 deficiencies, while CYP deficiency in 11B1 or 17‐hydroxylase deficiency results in mineralocorticoid excess. Finally, deficiency in the enzymes proximal to testosterone – StAR protein, CYP 17 (17,20‐desmolase), 17‐βHSD and 3‐ βHSD – results in under‐masculinization of males. Lastly, 3‐βHSD deficiency does not typically cause in utero virilization in females, because the steroid immediately above the enzyme block is dehydroepiandrosterone, a relatively weak androgen; however, mild postnatal signs of hypersecretion of this weak androgen, such as precocious pubarche, may be observed. Figure 8.2 Pathways of steroid synthesis in adrenal cortex. Shaded area indicates aldosterone synthesis in zona glomerulosa. Cortisol and androgen are synthesized in the zona fasciculata and zona reticularis, respectively. bHSD, b hydroxysteroid dehydrogenase. Aldosterone synthase, encoded by the CYP 11B2 gene, converts deoxycorticosterone (DOC) to aldosterone (Figure 8.3). Renin, secreted from the juxtaglomerular apparatus (JGA) in the kidney, causes cleavage of angiotensinogen into angiotensin 1, which is in turn cleaved by angiotensin‐converting enzyme (ACE) into angiotensin 2, a potent stimulator of CYP 11B2 activity, promoting aldosterone secretion from the zona glomerulosa. Renin will therefore be elevated when aldosterone synthesis is impaired, and suppressed with aldosterone excess. Aldosterone enters the distal renal tubular cells, binds to its receptor and enters the nucleus, causing enhanced activity of the Na+ K+ ATPase (‘the sodium pump’) and of the amiloride‐sensitive epithelial sodium channel (ENaC), resulting in sodium reabsorption. Figure 8.3 Schematic diagram of aldosterone stimulation and action. A, aldosterone; ACE, angiotensin converting enzyme; E, cortisone; ENaC, epithelial sodium channel; F, cortisol; JGA, juxtaglomerular apparatus; MR, mineralocorticoid receptor; 11βHSD2, 11‐betahydroxysteroid dehydrogenase 2. The following points should be noted: Glucocorticoid excess and deficiency states are often difficult to diagnose because cortisol levels normally fluctuate widely according to the time of day and degree of stress (Table 8.1). Glucocorticoid excess results in normal or high random cortisol levels, loss of the normal circadian rhythm, an increase in urinary free cortisol and failure of plasma cortisol to suppress with low‐dose dexamethasone. Table 8.1 Investigation of glucocorticoid secretion. Source: Reference values are taken from the Institute of Biochemistry, Glasgow Royal Infirmary. Salivary cortisol data kindly provided by Dr J. Schulga are derived from 147 healthy children aged 5–15 years. ACTH, adrenocorticotrophic hormone; CRH, corticotrophin‐releasing hormone. Glucocorticoid deficiency is easy to diagnose if the defect is in the adrenal gland itself (primary adrenal insufficiency) because this will result in elevated ACTH levels and low cortisol levels with absent or impaired rise following synthetic ACTH (Cortrosyn® or Synacthen®) stimulation. The diagnosis is more difficult when the defect lies in the hypothalamus or pituitary gland (central adrenal insufficiency, CAI). In suspected secondary adrenal insufficiency, the low‐dose synthetic ACTH test may be helpful. The rationale for this test is that the adrenal glands, if under‐stimulated for months or years, will respond subnormally to quasi‐physiological stimulation. A peak stimulated cortisol greater than 550 nmol/L is considered normal, while a peak of less than 400 nmol l−1 is considered too low, and a great many patients fall in the grey zone in between. We regard a peak‐stimulated cortisol level of >500 nmol l−1 as ‘normal’ (see Table 8.1) while recognizing that any chosen cut‐off level will be somewhat arbitrary and that the biochemical data must be interpreted in the clinical context. Mineralocorticoid deficiency is assessed by the measurement of plasma and urinary electrolytes, and of plasma renin and aldosterone (Table 8.2). Table 8.2 Investigation of mineralocorticoid secretion. AME, apparent mineralocorticoid excess; PHA, pseudohypoaldosteronism. a Aldosterone data obtained using Diagnostic Products Corporation Coat‐A‐Count Aldosterone Kit. b Plasma renin activity data obtained using BioChem ImmunoSystems Renin Maia Kit (Code 129640). Adrenal androgen status is assessed by plasma steroid analysis under basal and stimulated conditions (Table 8.3). Monitoring of patients with 21 hydroxylase deficiency is routinely based on the measurement of plasma 17‐hydroxyprogesterone (17‐OHP) and androstenedione at the time of clinic visits. If available, salivary or capillary profiles collected at home may add information about adherence to treatment. Table 8.3 Investigation of adrenal steroid precursors and androgens. Source: Blood spot 17‐hydroxyprogesterone data are from A.M. Wallace et al. (1986). Salivary data are from Dr Schulga (see Table 8.1). Plasma androstenedione and testosterone data are from M.G. Forest (1979). Cushing’s syndrome, unless iatrogenic, is very rare in childhood (Figure 8.4). However, its principal feature, obesity, is extremely common, so that the possibility of Cushing’s syndrome is often raised. Figure 8.4 A 10‐year‐old girl with Cushing’s disease causing growth failure and obesity. If a central cause for glucocorticoid excess or deficiency is discovered, imaging of the hypothalamic–pituitary area by magnetic resonance imaging (MRI) will be required. Adrenal ultrasound to detect adrenal enlargement is very operator‐dependent and computed tomography (CT) or MRI are needed to identify discrete lesions. The causes of Cushing’s syndrome in childhood are the following: The crucial difference between obesity, as a result of Cushing’s syndrome/disease, and exogenous obesity is in the growth pattern. Cushing’s syndrome/disease is almost always associated with a slower, and exogenous obesity with a faster, rate of linear growth. Appreciation of this difference in growth will largely protect children with simple obesity from being investigated for Cushing’s syndrome, although the occasional child with familial short stature and exogenous obesity may cause confusion. It is important not to subject children with simple obesity to investigation of Cushing’s syndrome since plasma and urine cortisol levels may be elevated in these subjects. Steroid biochemistry should also be interpreted with care in adolescent girls who may be on the oral contraceptive pill since this treatment increases CBG levels and hence plasma cortisol. Where exogenous steroid excess (by a steroid other than cortisol) is suspected, a low or undetectable 0800 hours plasma cortisol will usually confirm suppression of the pituitary–adrenal axis (see Table 8.1) and a low‐dose ACTH test is seldom necessary. If the features of Cushing’s syndrome are accompanied by virilization, then an adrenal tumour is likely and an adrenal CT scan should be performed promptly. The following can be carried out in a non‐specialist centre (from most to least practical): This can be difficult. Referral to a specialist centre is recommended. Iatrogenic Cushing’s syndrome must be managed by withdrawal of the glucocorticoid preparation or, if this is not possible, by minimizing the dosage given, or converting to an alternate day regimen. If severe adrenal suppression has occurred, hydrocortisone replacement may be required for months, or even years, following treatment. Treatment of an adrenal or pituitary tumour is by surgical resection, followed by hydrocortisone replacement if the other adrenal gland is suppressed. After bilateral adrenalectomy, mineralocorticoids should be given as well. Cushing’s disease is treated by trans‐sphenoidal surgical exploration with removal of the microadenoma. If none is identified, but petrosal sinus sampling has suggested lateralization of ACTH secretion, then a hemihypophysectomy can be carried out. A post‐operative cortisol level of <50 nmol/L [<1.4 μg/dL] indicates that the adenoma has been completely removed and the patient is cured. The treatment of choice for Cushing’s disease following failed pituitary exploration is direct pituitary irradiation. The alternative, bilateral adrenalectomy, carries a long‐term risk of Nelson’s syndrome (growth of the corticotroph adenoma). The ectopic ACTH syndrome may be seen as part of an established malignancy. Occasionally, small carcinoid lesions (e.g. lungs, pancreas) may be responsible, requiring location by CT scanning with selective venous sampling where necessary, and surgical removal. Cushing’s syndrome caused by bilateral micronodular dysplasia with or without primary pigmentation (Carney’s syndrome), or the McCune–Albright syndrome, should be treated medically if features are mild to moderate, otherwise by bilateral adrenalectomy. When surgical resection is impossible (e.g. with infiltrating carcinoma), or in pituitary Cushing’s disease when there may be a delay of several months before remission occurs (e.g. following radiotherapy), then medical therapy must be given. Useful agents include metyrapone and ketoconazole. In adrenal carcinoma, the adrenal cytotoxic o,p′‐dichlorodiphenyl‐dichloroethane (o,p′‐DDD) (mitotane) is of temporary benefit. After definitive treatment of Cushing’s syndrome, patients should be seen three‐monthly to monitor growth rate, body composition, and signs of recurrence. The causes of CAI are as follows: ACTH deficiency is usually seen in association with several other anterior pituitary hormone deficiencies. Severe ACTH deficiency at birth causes hypoglycaemia, poor feeding, convulsions and jaundice. The jaundice is of the cholestatic type and resolves once hydrocortisone is started. In older children, the symptoms of CAI are non‐specific. Cortisol deficiency is responsible for tiredness and lack of energy, with increased susceptibility to and a longer recovery period from minor illnesses. Children with POMC deficiency also have severe obesity and red hair. Some POMC mutations lead to high, not low, plasma ACTH. NFKB2 mutations are also associated with common variable immunodeficiency. In the newborn infant, dynamic studies are difficult to perform. If congenital hypopituitarism is strongly suspected (hypoglycaemia, jaundice, micropenis, low plasma free thyroxine (FT4) and random plasma cortisol levels less than 50 nmol/L [1.4 μg dL]), then replacement should be instituted without delay. In more doubtful cases where the child is reasonably stable, 1 μg ACTH can be given intravenously and the cortisol response assessed at 30 minutes. Peak values of <550 nmol/L [<19 μg/dL] are suggestive of prenatal hypostimulation of the adrenal glands. In children with suspected CAI, especially survivors of cancer, investigation depends on the severity of symptoms and the clinical context. Children who have received doses of >3000 cGy are particularly at risk. Cortisol secretion can be assessed by carrying out a low‐dose ACTH test, or a salivary cortisol profile. An undetectable plasma dehydroepiandrosterone sulphate (DHEAS) level at an age when adrenarche should have occurred (>8 years) is indirect evidence of at least partial ACTH deficiency. Hydrocortisone, 10 mg/m2 per day should be given divided in two or three doses per day. Treatment during an intercurrent illness or during surgery is as for 21‐hydroxylase deficiency (see section ‘Treatment’). Once the child is on glucocorticoid replacement, biochemical monitoring is not usually necessary. In the newborn period, cortisol deficiency will cause hypoglycaemia and jaundice, while aldosterone deficiency will result in hyponatraemia, hyperkalaemia, poor feeding, vomiting, and failure to thrive. Investigations will show elevated renin and ACTH, but low or normal 17‐OHP. Symptoms usually start from day 10 onwards. Boys with a DAX‐1 (dosage‐sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) mutation may have bilateral cryptorchidism in infancy and gonadotrophin deficiency in adolescence. Familial glucocorticoid deficiency (FGD, also known as ‘hereditary unresponsiveness to ACTH’) is an autosomal recessive disorder in which the adrenal cortex is unable to respond to ACTH. In about 25% of cases, a mutation on the ACTH receptor MC2‐R can be identified. These children, who tend to be tall, are classified as having FGD type 1. FGD without an MC2‐R mutation is termed FGD type 2. Recently, mutations in a new gene encoding a protein called melanocortin 2 receptor accessory protein (MRAP) have been identified in some cases of FGD type 2. MRAP is required for the expression of MC2‐R at the cell membrane. More recently identified causes include mutations in NNT (some with associated mineralocorticoid deficiency and insulin‐dependent diabetes) or in TXNRD2. Patients present with jaundice, poor feeding, failure to thrive, and hypoglycaemia in infancy. Older children may present with collapse and coma, sometimes with fatal consequences so that the diagnosis is only made post mortem. Investigations show elevated ACTH, low cortisol with poor or no response to ACTH and normal renin levels. Plasma potassium is usually normal but a degree of hyponatraemia may be present. This is attributable to cortisol deficiency causing impaired water secretion at the renal tubule. Congenital adrenal hypoplasia should be treated similarly to 21‐hydroxylase deficiency (see section ‘Treatment’), although the dose of hydrocortisone used can be somewhat lower (as there is no need to suppress ACTH to prevent hyperandrogenism). FGD should be treated with hydrocortisone but not fludrocortisone or salt. Autoimmune adrenalitis (Addison’s disease) is the most common cause of acquired primary adrenal deficiency. Although seen in isolation, it may also be associated with other autoimmune disorders, such as diabetes mellitus and Hashimoto’s thyroiditis, and with the polyglandular autoimmune (PGA) syndromes. PGA 1, also known as autoimmune polyendocrinopathy with endocrinopathy, muco‐cutaneous candidiasis and ectodermal dystrophy (APECED), is a recessive condition caused by mutations in the AIRE (autoimmune regulator) gene on chromosome 21 and has its onset in childhood and adolescence. Components of APECED syndrome include the following: ALD is an X‐linked recessive disorder affecting both the CNS and the adrenal cortex. Onset may be in childhood or early adulthood. The phenotype is highly variable, even within families, so that the presence and severity of neurological impairment and adrenal failure vary from case to case. Severely affected individuals suffer progressive neurological disability, and ultimately death. The neurological symptoms may precede those of adrenal insufficiency, or vice versa. Therefore, in all males with primary adrenal insufficiency other than CAH, plasma very long chain fatty acids must be measured. Symptoms include tiredness, weight loss, and, in the final stages, vomiting, drowsiness, and coma. Examination shows increased pigmentation (Figure 8.5), especially in the skin creases, old scars and buccal mucosa, with signs of recent weight loss, and low or normal blood pressure. Figure 8.5 Pigmentation and alopecia in a 10 year‐old‐girl who also had nail dystrophy caused by Candida. Investigations showed both Addison’s disease and hypoparathyroidism. Genetic studies showed a homozygous 13‐bp deletion in exon 8 of the autoimmune regulator gene on chromosome 21q22.3. Fasting glucose is normal or low. In acute adrenal insufficiency, sodium is low, potassium is high, and there may be severe hypoglycaemia. Basal cortisol is low or normal with no rise following ACTH administration. Plasma ACTH and renin activity are high. Given the association between Addison’s disease and other autoimmune diseases and ALD, the following is recommended at diagnosis and yearly thereafter: Acute adrenal insufficiency is managed by intravenous (IV) fluids with 0.9% saline and added 5% glucose, IV hydrocortisone (see p. 182) and conversion to oral hydrocortisone and fludrocortisone once recovery has occurred (Table 8.4). Table 8.4 Dosage schedules for mineralocorticoid, glucocorticoid and salt therapy in adrenal disorders. ACTH, adrenocorticotrophic hormone; CAH, congenital adrenal hyperplasia; FGD, familial glucocorticoid deficiency. Once a patient is on established treatment with hydrocortisone and fludrocortisone, clinical monitoring and adjusting the dosage as body surface area increases are usually sufficient and there is no need for laboratory investigations. Apart from the secondary hyperaldosteronism seen in cardiac and renal patients and in chronic volume depletion (leading to activation of the renin–angiotensin system), mineralocorticoid excess is very rare. Secondary hyperaldosteronism does not normally present to the endocrinologist. Fludrocortisone overdosage may result in hypertension with or without hypokalaemia. Carbenoxolone is rarely used, but liquorice addiction can cause the syndrome of AME, brought about by 11‐βHSD 2 inhibition (see below). Aldosterone‐secreting adrenal adenomas are rare in childhood, and other causes of hyperaldosteronism must be ruled out before invasive procedures, such as selective adrenal vein sampling, are performed. Two of the enzyme disorders causing CAH, 11‐hydroxylase and 17‐hydroxylase deficiencies, cause hypertension with hypokalaemia. 11‐hydroxylase deficiency causes virilization in girls and sexual precocity in boys from infancy, but affected children are hypertensive because of the high levels of DOC, rather than salt‐losing as in 21‐hydroxylase deficiency. 17‐hydroxylase deficiency causes cortisol and androgen deficiency but mineralocorticoid excess, so that affected 46,XY individuals are phenotypically female and may present with pubertal failure in association with hypertension and hypokalaemia. Deficiency of the enzyme 11‐βHSD 2 results in failure to metabolize cortisol to cortisone, with consequent occupation of mineralocorticoid receptors by cortisol (Figure 8.3). The clinical features are those of mineralocorticoid excess but aldosterone levels are suppressed, hence the term AME. Mutations of the CYP 11B2 gene may lead to a chimeric enzyme which is under ACTH control, resulting in glucocorticoid‐suppressible hyperaldosteronism. This condition should be suspected in the hypertensive adolescent, with or without hypokalaemia, in whom there is a family history of hypertension and cerebral haemorrhage. Treatment is with dexamethasone or amiloride. Liddle’s syndrome is autosomal dominant and caused by an activating mutation of the ENaC, leading to increased sodium absorption with aldosterone and renin suppression. Treatment is with amiloride. Mineralocorticoid deficiency causes salt‐wasting with polyuria, vomiting, dehydration, and hyperkalaemia which, if severe and untreated, leads to cardiac arrest. In older children, the salt‐wasting tendency is partially offset by an instinctive increase in salt intake. Congenital adrenal hypoplasia and Addison’s disease are discussed above; the three types of CAH causing salt‐wasting are discussed below. PHA may be associated with salt‐wasting and dangerous hyperkalaemia in the newborn period. Affected infants do not respond to large doses of fludrocortisone, and treatment consists of generous sodium replacement (Table 8.4) and resins to combat hyperkalaemia. The dominant and sporadic forms are milder and resolve with age. The autosomal recessive variety, caused by inactivating mutations in the ENaC, is severe and runs a protracted course. Isolated premature pubarche is common (see Chapter 5) but virilization in girls or prepubertal boys suggests an androgen‐secreting tumour or an enzyme defect. Virilization develops over a period of weeks or months and accelerated linear growth and bone maturation then become apparent. In boys, the testes are prepubertal (Figure 8.6). Figure 8.6 Penile enlargement, pubic hair development and scrotal laxity with prepubertal (2 ml) testes in a two‐year‐old boy. Investigation showed a small tumour in the right adrenal cortex. These show a marked elevation of one or more of the adrenal androgens, androstenedione, and DHEAS. Abdominal ultrasound, followed by CT or MRI scanning, should be obtained without delay. Treatment is by surgical removal wherever possible. If the tumour is well circumscribed and <5 cm in diameter, the prognosis is good. Pathological features are usually not helpful in predicting the risk of recurrence or of metastases. 21‐hydroxylase deficiency is by far the most common adrenal enzyme disorder and will be discussed in detail. 11‐ and 17‐hydroxylase deficiencies are discussed above (see p. 182). 3‐βHSD deficiency in females causes mild postnatal androgenization because of high DHEAS levels. Deficiency in P450 oxidoreductase, the electron donor for 21‐ and 17‐hydroxylases and aromatase, may be associated with bone abnormalities. In most populations, the incidence is about one in 15 000 births. 21‐hydroxylase deficiency results from mutations in the CYP21B gene, which is situated on chromosome 6. The phenotype of 21‐hydroxylase deficiency correlates reasonably well with the following genotype: SW 21‐OHD females present with virilization at birth. The clitoris is enlarged and there generally is posterior fusion of the labia majora (Figure 8.7). The vagina and urethra usually enter a common urogenital sinus, with the distal end of the vagina becoming increasingly more distant from the pelvic floor with the severity of virilization. Virilization can be graded in severity according to the Prader classification (Figure 8.8). Figure 8.7 Severe virilization with fusion of labia majora and gross clitoral enlargement in a female infant with 21‐hydroxylase deficiency. Figure 8.8 Prader classification of five stages of virilization in the female infant. Males with SW 21‐OHD usually present in the second week of life, with a salt‐losing crisis heralded by poor feeding, vomiting, poor weight gain, and listlessness. Examination shows dehydration with pigmentation of the scrotum. Biochemistry reveals hyponatraemia and hyperkalaemia. Death may occur before the diagnosis is considered. For this reason, neonatal screening for SW 21‐OHD is being implemented in an increasing number of countries. However, this is not yet universal because the positive predictive value of a high 17‐OHP is very low, especially in premature or stressed neonates. Boys with SV 21‐OHD, and girls with SV 21‐OHD in whom virilization at birth has been mild and/or missed, will present later in childhood (usually two to four years) with signs of androgen excess – long‐standing growth acceleration and advanced bone age, enlarged penis or clitoris, and pubic hair. Both girls and boys with SV 21‐OHD enter true puberty early when treated with glucocorticoids because of priming of the hypothalamic–pituitary axis. This leads to premature closure of the epiphyses and an adult height below target. NC 21‐OHD is a rare cause of hyperandrogenism in adolescent and adult females and should be considered in the differential diagnosis of the polycystic ovary syndrome. SW 21‐OHD is diagnosed in females with ambiguous genitalia, no palpable gonads, hyperpigmentation of the labia majora and a uterus on ultrasound, and in boys with a salt‐wasting crisis. Biochemical confirmation is based on high plasma androgens and 17‐OHP (values usually >100 nmol l−1 [>3300 ng dl−1]). In SV and NC 21‐OHD, 17‐OHP elevation is milder. Levels are higher between 8.00 a.m. and 9.00 a.m. In this context, 17‐OHP at 0 and 60 minutes after standard ACTH stimulation (250 μg), can be compared to the nomogram published by New et al. in 1983. After the birth of a child affected with 21‐OHD, the genotype of parents, affected child, and healthy siblings can be determined. The main purpose of prenatal testing is to assist with maternal dexamethasone treatment. Pre‐pregnancy counselling as to the pros and cons of this intervention is important. If the mother chooses to be treated, dexamethasone 20 μg/kg per day should be started by four to six weeks’ gestation. This should be continued throughout pregnancy if the foetus is an affected female, but stopped if the foetus is male or an unaffected female. Determining the sex of the embryo and CYP21B genotyping based on free embryonic DNA in maternal blood will allow shortening unnecessary treatments. The demonstrated benefit of reducing or preventing virilization in girls must be offset against hypertension, weight gain, striæ, and mood changes in the mother. The possible long‐term physical and psychological deleterious consequences of in utero dexamethasone exposure have led to this treatment no longer being even suggested to the couple by some clinicians. Diagnosis and management of 21‐OHD in newborn females are part of disorders of sex development (DSD) management (see Chapter 7). Once the diagnosis has been confirmed, treatment must be started as follows: If clinical evidence of salt‐wasting is present (weight loss >10%), manage as follows: Treatment consists of oral hydrocortisone, approximately 15 mg/m2 per day in three divided doses. Suspensions are unreliable and 10 mg hydrocortisone tablets should be given halved or quartered, asking the pharmacy to prepare powder if needed. Fludrocortisone 0.05–0.100 mg per day, with blood pressure monitoring. The dosage of 5 mmol/kg per day should be maintained for the first 12 months of life and is usually discontinued at, or shortly before, the first birthday. A great deal of time must be invested in counselling the parents at diagnosis. Parent information leaflets are very useful. Contact with a family with an affected child of similar sex can also be very helpful. Management of acute illness should be carefully discussed, and the parents instructed in the administration of intramuscular hydrocortisone in an emergency. The infant should be reviewed weekly until the parents are comfortable with management, thereafter one‐ to three‐monthly during infancy. Weight and length are measured on each occasion with blood pressure, plasma steroids, and renin at least three‐monthly. Routine measurement of electrolytes is not indicated if the child is thriving. Infancy is a prime time for acute illness, including gastroenteritis. The key to successful management is correct parental instruction (see Table 8.5), facilitated by provision of a CAH therapy card (similar to the asthma card). A CAH therapy card is shown in Appendix 2. In North America, it is common practice to prescribe a Medic‐Alert® bracelet to all patients who are glucocorticoid‐dependent. If the following procedure is followed, children with CAH rarely need admission to hospital and duration of hospital stay will be minimized: Table 8.5 Preoperative and emergency intramuscular (im), bolus, and infusion rate for hydrocortisone dosage (mg) according to body weight and age in months (m) and years (yr) in infants and children with adrenal insufficiency, including congenital adrenal hyperplasia. 12.5 mg for infants up to 6 mo, 25 mg from 6 mo to 5 yr, 50 mg from 5 to 10 yr, and 100 mg thereafter. Note that, should the iv line become obstructed or disconnected, cortisol deficiency will immediately ensue, making some centres prefer q6h hydrocortisone injections. On the day of surgery, hydrocortisone should be given intravenously at the doses stated in Table 8.5 as soon as an IV line is established. Thereafter, half of this initial dose should be given six hourly (see Table 8.5). This high dose hydrocortisone treatment also covers the mineralocorticoid needs of these patients. Chronic treatment consists of daily or twice‐daily fludrocortisone therapy to replace aldosterone, and twice‐ or thrice‐daily hydrocortisone to suppress ACTH secretion and thus prevent acceleration of bone maturation and virilization. The limitation of this approach is that the glucocorticoid regimen fails to mimic normal cortisol secretion. Moreover, even if ACTH secretion is normalized, the presence of the enzyme block will still cause androgen excess. Therefore, to achieve satisfactory adrenal suppression, a slightly supraphysiological dose of glucocorticoid is needed. Conversely, insufficient glucocorticoid dosage will result in androgen excess, early epiphyseal fusion, and decreased adult height. Central precocious puberty resulting from priming of the hypothalamus (see Chapter 5 on puberty) can be treated with gonadotrophin‐releasing hormone (GnRH) agonists but is not always easy to document with a GnRH stimulation test in children with CAH. It is also evident that frankly excessive glucocorticoid administration will result in Cushingoid features, obesity, and slow growth with decreased adult height. What is less obvious is that lesser degrees of glucocorticoid overdosage will cause hyperphagia resulting in obesity with normal or increased height velocity and advancing bone age, tempting the clinician to make a further increase in hydrocortisone dosage. With these considerations in mind, the clinician needs to make a careful judgement as to the optimal glucocorticoid dose, taking the clinical assessment, height velocity, bone age, and biochemical profile into account, and being mindful that adherence problems are common, especially during adolescence. Hydrocortisone – 10–15 mg/m2 per day –is usually given in two to three doses, titrating the dosage so that the height velocity is 4–7 cm per year. The manner in which the daily glucocorticoid dose should be administered is controversial. Some favour giving a thrice‐daily dose, equally divided, in pre‐school children, then a twice‐daily dose from school age (when adherence with a thrice‐daily regime becomes problematic). Fludrocortisone should be given at 0.150 mg/m2 per day to bring plasma renin into the reference range. At clinic visits (no less than every six months), the following should be done: The sources of morbidity for parents and individuals with CAH are as follows: The principal causes of morbidity are the complications of surgery in girls, and of obesity in both sexes. Strategies to minimize these problems include the use of prenatal dexamethasone, bilateral adrenalectomy during infancy in those with SW 21‐OHD associated with severe genotype, adding growth hormone, anti‐androgens and aromatase inhibitors, or using sustained‐release hydrocortisone. However, none of these options has become standard, despite having been under discussion for many years. Feminizing adrenal adenomas are exceedingly rare. The clue that the source of sexual precocity lies outside the hypothalamic–pituitary axis is that luteinizing hormone (LH) and follicle stimulating hormone (FSH) are undetectable and do not respond to GnRH. The androgen deficiency in boys with adrenal hypoplasia is insignificant because gonadal androgen synthesis is intact. However, severe enzyme deficiency affecting both adrenal and gonadal androgen synthesis results in under‐masculinization with complete sex reversal in the more severe cases, and ambiguous genitalia in less severe cases. Management is discussed in Chapter 7. Oestrogens are responsible for closure of the epiphyses at adolescence. Gene mutations inactivating the aromatase enzyme or of the oestrogen receptor are associated with tall adult stature. Chromaffin cell tumours within the adrenal are called phaeochromocytomas while if extra‐adrenal they are called paragangliomas. Phaeochromocytoma and paragangliomas (PPGLs) may also occur in association with neurofibromatosis type 1 or as part of the multiple endocrine neoplasia (MEN) syndromes: MEN type 2A (hyperparathyroidism, phaeochromocytoma, medullary thyroid carcinoma), MEN type 2B (phaeochromocytoma, medullary thyroid carcinoma, and tongue and lip neuromas), von Hippel–Lindau syndrome or renal and gastrointestinal tumours in succinate dehydrogenase deficiency. All these may occur sporadically or as dominantly inherited disorders. Although extremely uncommon in childhood, PPGLs are an important cause of hypertension. It is suggested by the combination of episodic pallor, sweating, and headaches, with either sustained hypertension or raised blood pressure during a symptomatic episode. The diagnosis is made by examining plasma metanephrines in children with hypertension and in children with symptoms suggestive of a PPGLs, even in the absence of hypertension. Neuroblastoma usually presents to the general paediatrician or oncologist. The diagnosis is suggested by hypertension in the context of malaise, sweating, pallor, and an abdominal mass. There is an increase in catecholamine metabolites in the urine. NB: Involvement of a specialist centre does not preclude a shared care arrangement. Correct emergency management depends on: (i) recognizing adrenal insufficiency in an acute situation; and (ii) giving appropriate glucocorticoid, glucose, electrolyte and fluid treatment to known or suspected cases. The cardinal symptoms include vomiting, diarrhoea (often mistakenly thought to be infectious in nature) and altered consciousness, and with/without a background history of poor feeding, poor weight gain/weight loss. Signs of adrenal insufficiency include dehydration, shock, hyperpigmentation (in primary adrenal insufficiency), and genital anomaly (in genetic females with 21‐hydroxylase deficiency). Investigation suggesting adrenal insufficiency includes hypoglycaemia, hyponatraemia (due to salt‐wasting from aldosterone deficiency or to impaired free water clearance due to cortisol deficiency), hyperkalaemia, and acidosis. In the newborn, adrenal insufficiency causes hypoglycaemia, prolonged jaundice and, sometimes, a neonatal hepatitis‐like syndrome with conjugated bilirubinaemia and raised liver transaminases. There may be associated features such as roving nystagmus (in septo‐optic dysplasia) and low T4 with low or inappropriately normal TSH in hypopituitarism. It is particularly important to consider adrenal insufficiency in a newborn infant with unexpected hypoglycaemia, for example, when neither the gestation nor birth weight explains this problem, and especially if there is concurrent jaundice. Hospital admission is almost always required in a patient with known adrenal insufficiency who develops vomiting (see Table 8.1). Such patients should always be seen at the hospital, and only allowed to leave the emergency department after a senior member of staff has been consulted.
Adrenal Disorders
Physiology
Adrenal anatomy and physiology
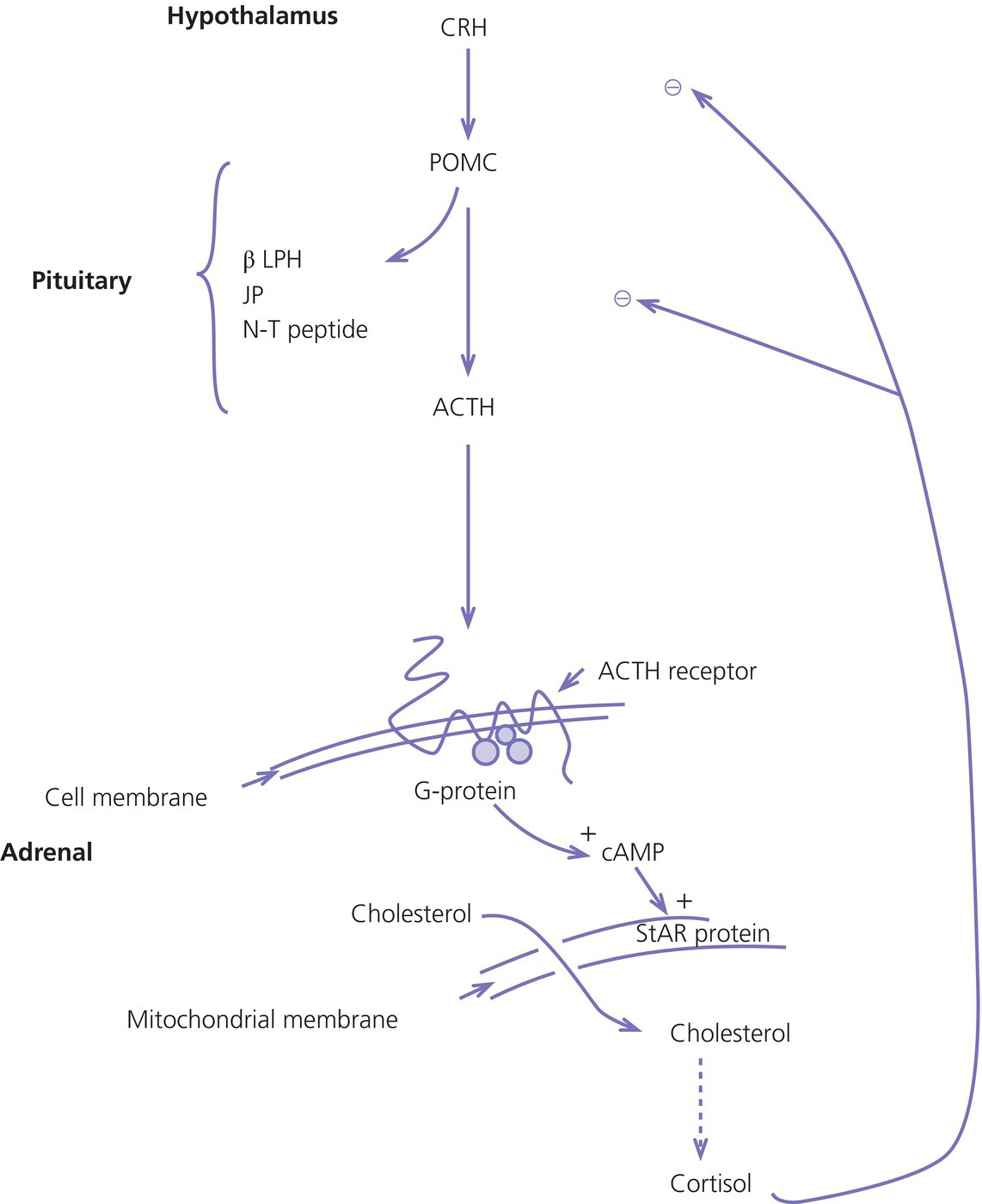
Hypothalamic–pituitary axis
Steroid pathways
Mineralocorticoid synthesis
Investigations of adrenocortical function
Biochemical
Glucocorticoid secretion
Basal samples
Reference values
Plasma
Fasting glucose
4–7 mmol/L
0800 h ACTH
<20 mU/L
2400 h (sleeping) cortisol
<50 nmol/L
0800 h cortisol
200–700 nmol/L
Saliva
8 a.m. cortisol
6.5–28 nmol/L (usually 9–16) median 14.5
6 p.m. cortisol
≤10 (mean 1.4)
Urine
Urinary‐free cortisol (random)
<25 μmol/mmol/L creatinine
Suppression tests
Low‐dose dexamethasone (1 mg at 11 p.m.)
Plasma cortisol <50 nmol/L at 8 a.m. the next day
High‐dose dexamethasone (2 mg 6‐h × 8)
Plasma cortisol <50% basal value 48 h after last dose
Stimulation tests
CRH (1 μg/kg IV)
Doubling of ACTH (usually to 8–25 mU/L)
Cortisol peak ≥600 nmol/L with >20% increase over basal value
Standard short Synacthen (250 μg m−2 IV)
Peak cortisol >500 nmol/L± doubling of basal value
Low‐dose short Synacthen (500 ng m−2 IV)
Peak cortisol >500 nmol/L± doubling of basal value
Insulin hypoglycaemia (min blood glucose <2.2 mmol/L)
Peak cortisol >500 nmol/L
Metyrapone test
Doubling of plasma 11 deoxycortisol and urinary tetrahydrodeoxycortisol
Mineralocorticoid secretion
Electrolytes
Normal values
Mineralocorticoid excess
Mineralocorticoid deficiency
Sodium
Na 135–145 mmol/L
Normal or ↑
↓
Potassium
K 3.5–4.5 mmol/L
Normal or ↓
↑
Aldosterone (ρmol l−1)a
Infants <1 mo
1000–5500
↑ in hyperaldosteronism
↓ in hypoaldosteronism
Infants 1–6 mo
500–4500
↓ in AME and Liddle’s syndrome
↑↑ in PHA
Infants 6–12 mo
160–3000
Children 1–4 yr
70–1000
Children 5–15 yr
30–600
Adults
30–420
Plasma renin activity (ng (ml/h)b (supine for 30 min)
Infants (<1 yr)
<31
Suppressed
↑
Children 1–4 yr
<26
Children 5–15 yr
<9
Adults
<2.6
Adrenal androgen secretion
17‐hydroxyprogesterone
Plasma (nmol l−1 )
Normal neonates
<13
Stressed/preterm neonates
<40
Neonates with CAH
>100
Adults
Basal
<13
60 min after 250 μm iv Synacthen
<20
Blood spot (nmol/L)
Extraction assay
Direct assay
Stressed/preterm neonates
<70
<140
Neonates with CAH
>180
>350
Saliva (nmol/L of saliva)
Children 5–15 yr 09.00 h
0.16–0.66 (median range 0.22–0.4)
Androstenedione (mean ± SD)
Male
Female
Plasma (nmol/L)
1–2 mo
1.5 ± 0.45
0.66 ± 0.24
>6 mo–adrenarche
<0.4
<0.4
Adult male
3.7 ± 0.9
Adult female
follicular phase
2.7 ± 1.0
luteal phase
5.2 ± 1.5
Saliva (nmol/L)
5–15 yr 09.00 h
0.04–0.96 (median range 0.2–0.7)
Testosterone (mean ± SD)
Male
Female
Plasma (nmol/L)
First wk
1.15 ± 0.15
0.45 ± 0.3
1–2 mo
8.8 ± 2.7
0.28 ± 0.13
>6 mo–prepubertal
<0.3
<0.3
Adult male
19.8 ± 4.7
Adult female
follicular phase
0.75 ± 0.2
luteal phase
1.28 ± 0.3
Dehydroepiandrosterone sulfate (μmol/L of plasma)
Male
Female
Infancy, pre‐adrenarche
<2
<2
Adult
2–9
2–11
Glucocorticoid excess
Cushing’s syndrome
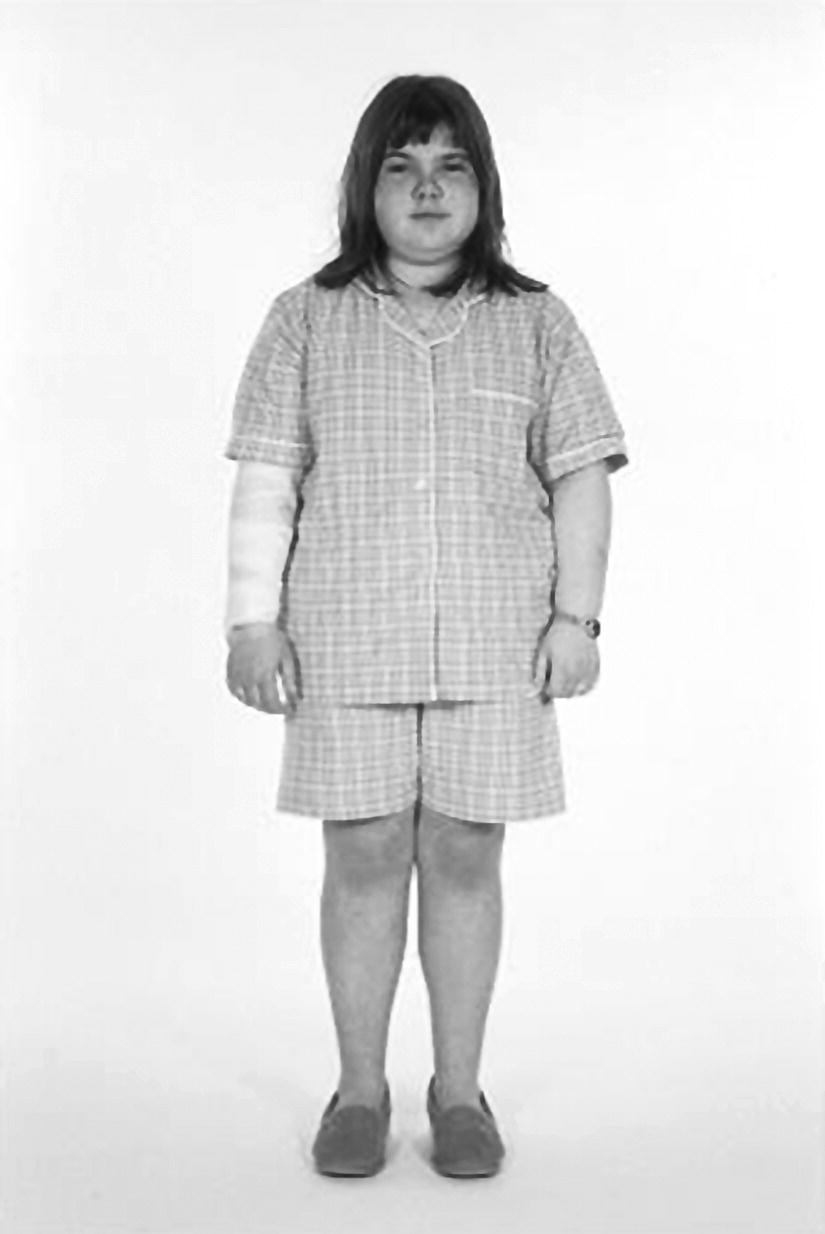
Imaging
Causes
Clinical diagnosis
Symptoms
Signs
Growth
Investigations
Diagnosis of Cushing’s syndrome or disease
Aetiological diagnosis of hypercortisolism
Treatment
Follow‐up
Glucocorticoid deficiency
Adrenal insufficiency caused by hypothalamic–pituitary disease – central adrenal insufficiency (CAI)
Causes
Clinical features
Investigations
Treatment
Primary adrenal insufficiency
Causes
Congenital adrenal hypoplasia
Familial glucocorticoid deficiency
Treatment
Acquired adrenal insufficiency
Causes
Clinical features of primary adrenal insufficiency
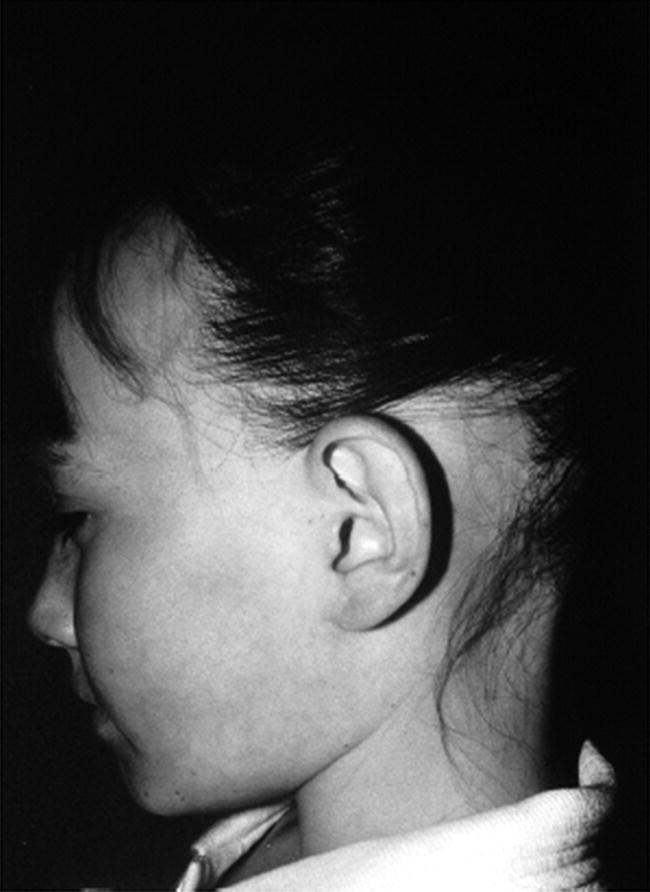
Investigations
Treatment
Disorder
Mineralocorticoid
Glucocorticoid
Salt
Hypothalamo–pituitary‐adrenal insufficiency
Not required
Hydrocortisone 8–10 mg m2/day
Primary adrenal insufficiency (Addison’s disease)
Fludrocortisone 100–200 μg/day:
Hydrocortisone 10–12 mg/m2/day
Congenital adrenal hypoplasia
As for primary adrenal insufficiency
For first 6 mo consider treatments as for CAH. (see p. 190), then hydrocortisone 10–12 mg/m2/day
FGD
Not required
As for primary adrenal insufficiency (Addison’s disease)
CAH (21‐hydroxylase deficiency)
Salt‐wasting
As in Addison’s
For first 6 mo of life, see pp. 195. Oral hydrocortisone 10–15 mg mg/m2/day thereafter.
5 mmol/L/kg/day in three divided doses for first yr
Dexamethasone 0.25 mg hs after epiphyseal fusion (check for iatrogenic Cushing’s)
Simple virilizing
As above if renin ↑ at diagnosis
As above
Not required
Pseudohypoaldosteronism
Not usually effective
Sodium chloride 10–40 mmol kg/day+ calcium or sodium resonium
Follow‐up
Mineralocorticoid excess
Causes
Mineralocorticoid deficiency
Causes
Aldosterone deficiency
Aldosterone resistance (pseudohypoaldosteronism)
Clinical features and investigation
Sex steroid excess
Androgen excess
Adrenal causes of androgen excess
Adrenocortical tumour
Clinical features
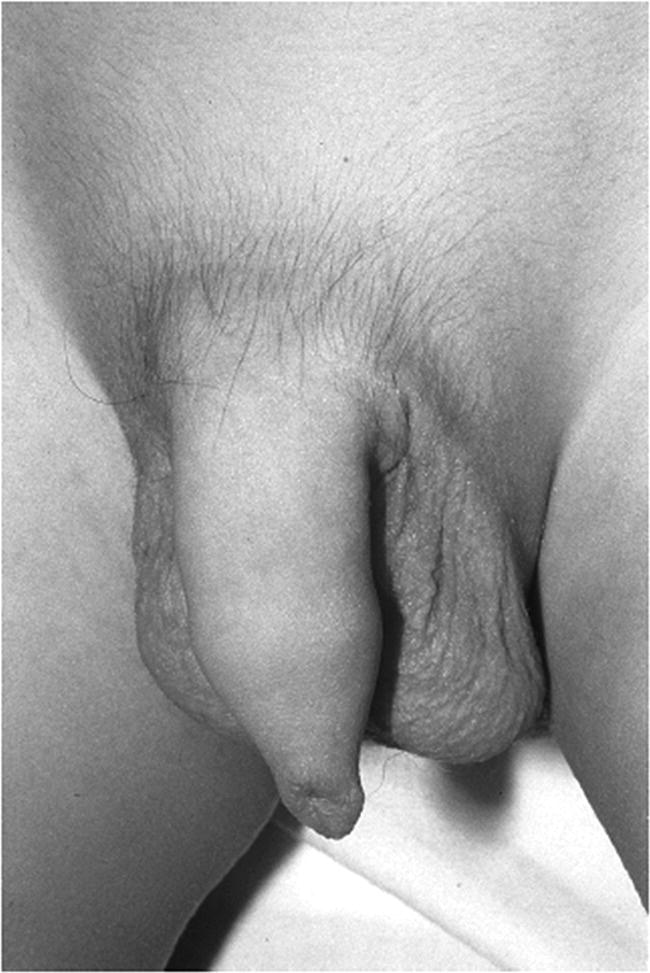
Investigations
Treatment
Adrenal enzyme defects
21‐hydroxylase deficiency
Incidence
Pathogenesis and classification
Clinical features
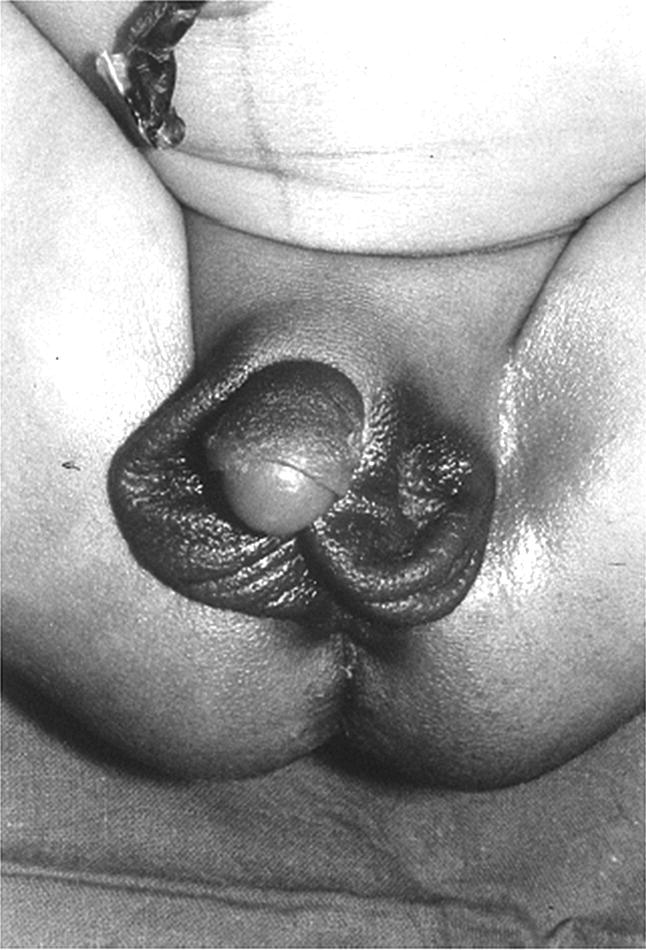
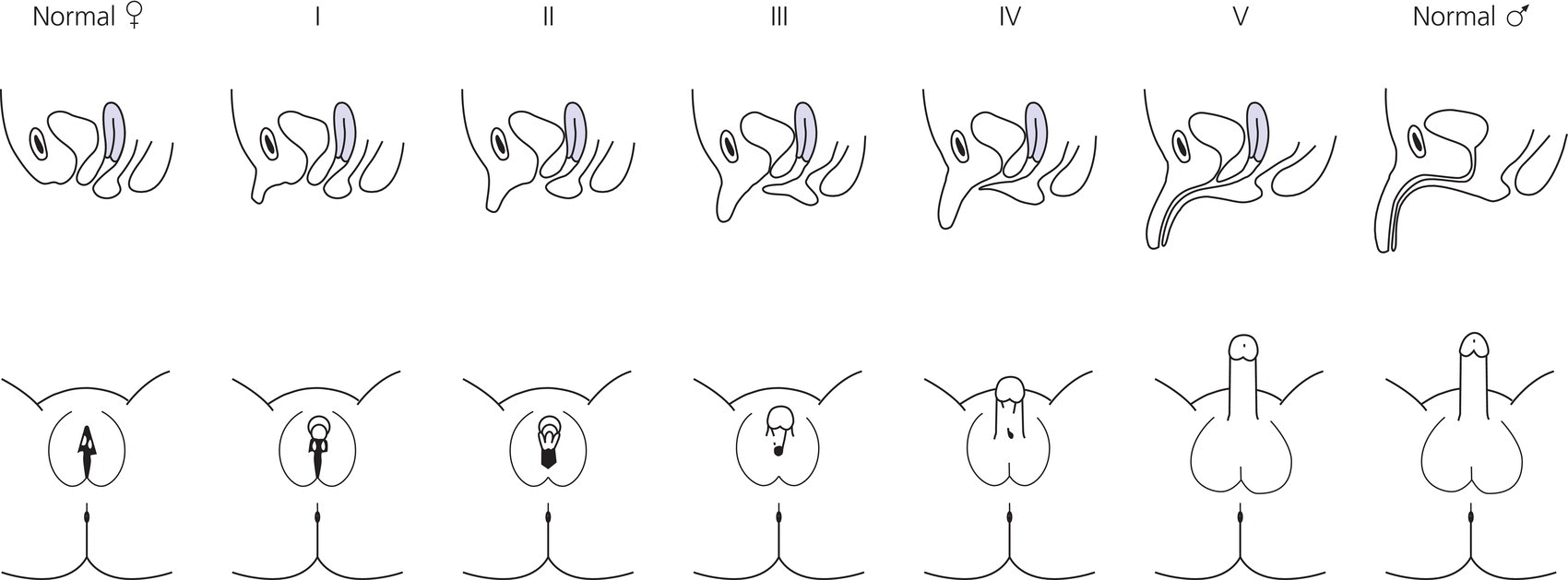
Diagnosis
Prenatal management
Neonatal management
Management in infancy
Glucocorticoid Therapy
Fludrocortisone Therapy
Salt Therapy
Education and Surveillance
Management of Acute Illness
Subsequent 6h infusion rate
Age
Weight (kg)
im dose
Bolus dose
mg h−1
<6 m
3–7
12.5
6.25
0.5
6 m–5 yr
7–20
25
12.5
1
5–10 yr
20–30
50
25
2
10 yr
30
100
50
3
Medical Management of Children Undergoing Surgery
Management of Linear Growth
Morbidity
Oestrogen excess
Sex steroid deficiency
Adrenal androgen deficiency
Causes
Oestrogen deficiency
Adrenal medullary disorder
Phaeochromocytoma and paraganglioma (PPGL)
Neuroblastoma
Future developments
Potential pitfalls
Controversial points
When to involve a specialist centre
Transition
Emergency management
Recognizing adrenal insufficiency
Appropriate management of adrenal insufficiency
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


