Adrenal Cancer
 ANATOMY
ANATOMY
The paired suprarenal (adrenal) glands are located between the superomedial aspects of the kidney and the diaphragmatic crura. They are surrounded by connective tissue containing perinephric fat. The glands are enclosed by renal fascia, but separated from the kidneys by fibrous tissue. The triangular right gland relates to the diaphragm posteriorly and the inferior vena cava and liver anteriorly. The semilunar left adrenal gland is positioned in the middle of the left crux of the diaphragm. The omental bursa separates it from the stomach. It is also related to the spleen and pancreas.1
The endocrine function of the adrenal glands necessitates an abundant blood supply. The superior suprarenal arteries are derived from the inferior phrenic artery, the middle suprarenal arteries from the abdominal aorta near the origin of the superior mesenteric artery, and the inferior suprarenal arteries from the renal artery. A large central vein leaves the anterior surface of the gland at the hilum. The shorter right suprarenal vein drains into the inferior vena cava and the longer left suprarenal vein drains into the left renal vein.1
The lymphatic drainage follows the arterial supply and is predominantly to lumbar lymph nodes. The superior lymphatic trunks end in aortocaval lymph nodes located near the origin of the celiac plexus. The inferior lymphatic trunks end in lateroaortic nodes above the renal pedicle. Some trunks may pass through the diaphragm, following the splanchnic nerves, ending in retroaortic nodes in the posterior mediastinum. On the right, some lymphatic trunks may penetrate the liver.2
The adrenal gland is composed of a central catecholamine-producing medulla enveloped by the steroid-secreting cortex. Although they are in intimate contact, they represent two functionally separate organs with different embryologic origins.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Adrenal Cortical Tumors
Adrenocortical tumors are rare. Benign tumors are more common, occurring in 1% to 8% of the general population, and the incidence of carcinoma is approximately 1 per million population in the United States.3,4 There are approximately 75 to 115 new cases per year.5 Adrenal cortex carcinoma deaths account for 0.2% of all yearly cancer deaths. There is a bimodal age distribution with disease peaks before the age of 5 years and in the fourth to fifth decades of life.
Overall, adrenocortical carcinoma (ACC) is slightly more common in women than men. Nonfunctional carcinomas occur in an older age population (>30 years old) and are more common in men (3:2 male-to-female ratio), although functional tumors are more common in women (7:3 female-to-male ratio) and younger patients. As they frequently present with symptoms related to hormone production, functional tumors are usually detected at an early stage.
Although most cases of ACC are sporadic, it has been described as a component of several hereditary cancer syndromes, including Li-Fraumeni syndrome (breast cancer, soft tissue and bone sarcoma, brain tumors, and ACC), Beckwith-Wiedemann syndrome (Wilms’ tumor, neuroblastoma, hepatoblastoma, and ACC), multiple endocrine neoplasia type I (parathyroid, pituitary and pancreatic neuroendocrine tumors, and adrenal adenomas and carcinomas), and SBLA syndrome (sarcoma, breast, lung, ACC, and other tumors).6–9 A role for p53 mutations in sporadic ACC is suspected.
Adrenal Medulla Tumors
Ganglioneuromas are rare, benign tumors of the adrenal medulla seen in children and young adults.10,11 Neuroblastoma is the most common malignant tumor of the adrenal gland in children, accounting for 90% of all cases.3,12
Pheochromocytomas and functional ganglioneuromas (or extra-adrenal pheochromocytomas) are rare tumors that rise from chromaffin cells in the adrenal medulla and elsewhere. They secrete catecholamines and cause intermittent, episodic, or sustained hypertension. Pheochromocytomas have an estimated prevalence of 0.1% in hypertensive patients.13 In autopsy series, there is a 0.01% to 0.1% prevalence of unsuspected pheochromocytomas. Extra-adrenal tumors are more commonly malignant.14 Estimates of the incidence of malignancy in pheochromocytoma range from 5% to 46% in different series.15,16 Approximately 400 new cases of malignant pheochromocytomas are expected each year in the United States.17,18
Pheochromocytomas may be associated with a variety of endocrine and nonendocrine inherited disorders. Bilateral pheochromocytomas are a component of multiple endocrine neoplasia type IIa (MEN-IIA) syndrome (pheochromocytoma, medullary thyroid carcinoma, and parathyroid hyperplasia) or MEN-IIB syndrome in which they are associated with marfanoid habitus, mucosal neuromas, and medullary thyroid carcinoma. Pheochromocytomas occur in 25% of patients with von Hippel-Lindau syndrome and <1% of patients with neurofibromatosis and von Recklinghausen’s disease.19,20
 NATURAL HISTORY
NATURAL HISTORY
Fifty-nine percent of adrenal cortex tumors are functional, the left-to-right ratio is approximately 1:1, and 2.4% are bilateral.21 Diagnosis is frequently delayed because of the rarity of disease and the deep retroperitoneal location of the adrenal glands.22
Nonfunctioning ACCs are typically larger tumors, >6 cm, while functioning tumors tend to be discovered at an earlier stage (for TNM staging, see Table 76.1). Incidentally discovered adrenal masses <3 cm are rarely malignant. ACC is an aggressive malignancy that frequently violates the tumor capsule and invades surrounding tissues. It metastasizes to lungs, liver, brain, and regional lymph nodes. Many patients present with widespread metastasis; most of these patients die within 6 months of diagnosis. This situation is especially common in the pediatric population. For all stages, the 5-year overall survival is only 20% to 25%.22
Malignant pheochromocytomas exhibit a similar pattern of spread but also metastasize to bone. They are equally common in men and women. The average age of presentation is 40 to 50 years old, but these carcinomas may also occur in children.
TABLE 76.1 TNM STAGING FOR ADRENOCORTICAL CARCINOMA

 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Functional adrenocortical tumors most frequently secrete cortisol and androgens, resulting in Cushing syndrome, virilization, and hypertension. Estrogen or aldosterone production is less common. Approximately 60% of ACCs are functional. In a child, virilization is the most common symptom of ACC, and Cushing syndrome is relatively uncommon. By contrast, adults usually present with either Cushing syndrome alone or mixed with virilization. Patients with nonfunctioning tumors present with nonspecific symptoms related to tumor burden, including abdominal fullness, early satiety, pain, weight loss, weakness, fever, or an abdominal mass. Nonfunctioning ACCs are more common in older patients and tend to progress more rapidly. An increasing number of adrenal tumors are incidentally discovered during abdominal imaging in the absence of any symptoms.
Pheochromocytomas arising in the setting of an inherited disorder occur in the adrenal medulla in 90% of cases, as compared with 75% of sporadic pheochromocytomas. When associated with MEN-II syndromes, 80% are bilateral. A tumor >5 cm more commonly has a malignant course than a smaller lesion. Serum catecholamines and urinary metanephrine and vanillylmandelic acid levels are elevated in 90% of pheochromocytomas. These patients present with a range of symptoms from mild labile hypertension to sudden cardiac death secondary to hypertensive crisis, myocardial infarction, or cerebrovascular accident. The classic triad of symptoms consists of episodic headaches, diaphoresis, and tachycardia.8,23 About half of patients have paroxysmal hypertension, and others have sustained hypertension. Pheochromocytomas may also present with normal blood pressure in 5% to 15%. Other symptoms may include pallor, palpitations, panic attack symptoms, or generalized weakness. Orthostatic hypertension may occur in association with hypovolemia.
FIGURE 76.1. T2-weighted magnetic resonance image of a 35-year-old woman with a left adrenal carcinoma.
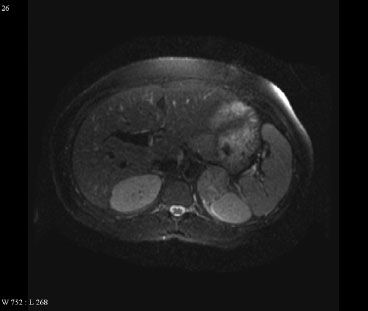
TABLE 76.2 DIAGNOSTIC WORKUP FOR ADRENAL TUMORS

TABLE 76.3 CLASSIFICATION OF ADRENAL TUMORS

 DIAGNOSTIC WORKUP AND STAGING
DIAGNOSTIC WORKUP AND STAGING
Patients with adrenal gland tumors should be evaluated for other primary tumors, because metastasis to the adrenal gland is common. In metastatic tumors that are not adrenal in origin, a biopsy is recommended. Prior to obtaining a biopsy, however, pheochromocytomas must be ruled out by measuring the fractionated plasma-free metanephrine, as well as the 24-hour urine fractionated metanephrines and catecholamines. Furthermore, any patient with a suspected adrenal tumor should be screened for hormonal hypersecretion, including cortisol, aldosterone, and catecholamine secretion (Table 76.2). As hypercortisolism is the most frequent abnormality, serum cortisol, a 24-hour urinary cortisol, and an overnight dexamethasone suppression test should be obtained.
Morphologic evaluation of the adrenal glands with computed tomography (CT) of the abdomen should be performed as per adrenal protocol. CT of the abdomen with thin cuts through the adrenal gland is the imaging test of choice for the evaluation of adrenal tumors. Carcinomas can mimic adenomas but are characterized by larger size, irregular margins, and heterogenous enhancement. They may also demonstrate tumor necrosis and cystic degeneration. Local invasion, tumor extension into the vena cava, as well as lymph node or other metastases are often seen in advanced ACC. Magnetic resonance imaging (MRI) is useful in the evaluation of adrenal tumors (Fig. 76.1). ACC and pheochromocytomas are hyperintense on T2-weighted images, and venous invasion is better imaged on MRI. If there is still question of vascular invasion, an angiographic study can be performed preoperatively, either selective arteriography or vena cavography. Fluorodeoxyglucose positron emission tomography may be useful in differentiating benign from malignant lesions. It may also serve as an additional staging study for patients with known ACC. A chest CT should also be performed.
The diagnosis of pheochromocytoma is confirmed by measurements of urinary and plasma fractionated metanephrines and catecholamines. Medication-induced elevation of metanephrine levels is common and should be evaluated prior to initiating further costly workup. Levels of metanephrine 4 times the normal limit are diagnostic of pheochromocytoma, and biochemical confirmation of disease is followed by imaging to locate the tumor. Other scans, including the OctreoScan (Mallinckrodt Inc., St. Luis, MO) and a bone scan, may be indicated if metastatic disease is suspected.
Levels of plasma aldosterone (high) and renin (low) activity should be assessed when suspecting primary aldosteronism (hyperaldosteronism). The ratio of aldosterone to renin above 30 is suggestive of hyperaldosteronism and should be confirmed with saline suppression or salt loading tests, and electrolytes should be measured. Although malignant hyperaldosteronism is rare, these patients require an open adrenalectomy to prevent tumor rupture. In benign cases of hyperaldosteronism, adrenal vein sampling for aldosterone is considered standard of care.
Patients with Cushing syndrome require an evaluation of serum levels of corticotropin, cortisol, as well as the sex steroid dehydroepiandrosterone sulfate (DHEA-S). If cortisol levels are elevated, a confirmatory test with dexamethasone suppression is recommended. It is important to remember that elevated corticotropin levels do not indicate adrenal origin, and further workup is necessary to look for primary causes of increased corticotropin. Benign adrenal tumors can be removed laparoscopically. Patients who present with Cushing syndrome in the presence of large (>5 cm), inhomogeneous, or invasive masses should be suspected to have malignant tumors, and imaging of the chest, abdomen, and pelvis is warranted to look for metastasis.
Pathologic Classification
Tumors <6 cm are more likely adenomas, although some smaller tumors may be malignant (Table 76.3). Hemorrhage and necrosis may be observed macroscopically in carcinomas. Numerous mitotic figures and cellular undifferentiation are common microscopic findings. Larger size, vascular invasion, or invasion of surrounding tissues and numerous mitotic figures are poor prognostic features.24
Pheochromocytomas have malignant features in <10% of cases. Macroscopically, they tend to be encapsulated with areas of cystic change, hemorrhage, and necrosis. The capsule is frequently invaded, but that does not constitute malignant change. Benign and malignant pheochromocytomas may appear identical histologically. The only absolute criterion for malignancy is metastasis.9 Histologically, cell size, nuclear size, and arrangement of cells are variable. A twisted cell cord pattern, basophilic or cytophilic staining with fine intracytoplasmic pigment granules, and periodic acid-Schiff staining of secretory droplets aid in the diagnosis.
 GENERAL MANAGEMENT
GENERAL MANAGEMENT
Benign tumors are nonfunctioning and are most often found incidentally. After the benign nature of these tumors is confirmed, small tumors can be left untreated and can be followed up by repeat imaging 6 to 12 months after diagnosis to confirm their stability. Larger tumors (>4 cm) should be followed up with imaging in 3 to 6 months after diagnosis. A small benign mass that grows at the rate of >1 cm per year can be removed electively, but any significant growth of a larger mass should alert the clinician to a potentially malignant nature of the tumor and adrenalectomy is recommended in that case.
Nonfunctioning adrenal tumors that are >4 cm, are heterogenous, or have irregular margins are suspicious for adrenal carcinomas. Surgery with removal of adjacent lymph nodes is the primary treatment for ACC. Complete resection is the only treatment that offers long-term disease-free survival, but it is not always feasible. For patients with a macroscopically complete resection, a margin-free resection is a strong predictor for survival. Efforts to avoid tumor spillage are warranted, and the tumor capsule should remain intact. Invasion or adherence of adjacent structures often necessitates en bloc resection of the kidney or spleen, partial hepatectomy, or pancreatectomy. The presence of tumor thrombus in the renal vein or vena cava does not preclude resection. A lymphadenectomy is often included. The role of tumor debulking in the presence of metastatic disease is not clear. Incomplete resection of the primary tumor or metastatic disease not amenable to surgery is associated with a poor prognosis. Still, tumor debulking may help control hormonal oversecretion or relieve local symptoms in certain cases. Even with a complete resection, local recurrence and metastatic disease are common. For isolated distant metastases and locally unresectable disease, cytoreductive resection with or without radiation is recommended.
The role of radiation in the management of ACC is not well defined. It has been proposed as adjuvant therapy in high-grade adrenal carcinoma as well as after complete resection or as management of microscopic residual disease. One series reports a 10-year crude survival rate of 33% for surgical resection followed by adjuvant radiation.25 External radiation results in good response rates and effective palliation in patients with residual macroscopic disease or bone or nodal metastasis.18,26
Mitotane, a chemical congener of the insecticide DDT (dichlorodiphenyltrichloroethane), is an adrenolytic compound with specific activity on the adrenal cortex. It is the chemotherapeutic agent most commonly used in the management of ACC. In patients with measurable disease, overall response rates of 14% to 36% have been reported, but most studies have reported no significant survival benefit.27 The largest retrospective study from Italy and Germany that included 177 patients with resected ACC (stages I to III) showed improvements in disease-free survival and overall survival.28 Unfortunately, responses are usually partial and transient, with only an occasional complete remission.21 The role of mitotane as adjuvant therapy after complete surgical resection is questionable. Despite limited supporting data, it is frequently employed in this setting, given the high rates of locoregional and distant recurrence. Serum levels of mitotane are monitored in order to optimize therapy because objective response in the metastatic setting was associated with higher serum levels (>14 mg/L). Unfortunately, increased toxicity is also associated with higher serum levels. Side effects are predominantly gastrointestinal, particularly nausea, but anorexia and diarrhea also occur. Although less common, central nervous system toxicity can include lethargy, somnolence, ataxia, dizziness, or confusion.
Single-agent chemotherapy has proven disappointing in the management of ACC. Doxorubicin and cisplatin have both been evaluated as single-agent therapy and in combination with mitotane. Neither drug was efficacious.29 Multiagent chemotherapy has shown more promise. A multicenter phase II study by the Italian Group for the Study of Adrenal Cancer demonstrated 49% overall response rate using a regimen of etoposide, doxorubicin, and cisplatin in combination with mitotane. The regimen was well tolerated. The most common side effects were gastrointestinal. The time to progression in responding patients was 2 years.6 Inclusion of mitotane in a multidrug regimen is rational as ACCs are prone to multidrug resistance mediated by the multidrug resistance-1/P glycoprotein drug pump, whose mechanism is inhibited by mitotane. This multidrug regimen is worthy of further study.
Surgical resection is the definitive management of pheochromocytoma, but it is a high-risk procedure. Cardiovascular and hemodynamic parameters must be monitored closely. Preoperative medical therapy is aimed at controlling hypertension and expanding intravascular volume. Preoperative pharmacologic preparation typically includes combined α– and β-adrenergic blockade.
In patients with undiagnosed pheochromocytomas who undergo surgery for other reasons, surgical mortality rates are high because of lethal hypertensive crisis and multiorgan failure.30 In the largest series of 147 patients undergoing surgery for pheochromocytoma, perioperative mortality and morbidity rates were 2.4% and 24%, respectively.31 Although it results in rapid symptomatic control, surgical removal of a pheochromocytoma does not always lead to a long-time cure. In a large series of 176 patients, pheochromocytoma recurred in 16% of patients. Half of these recurrences were malignant.32 In patients with bilateral pheochromocytomas, usually in the setting of an inherited syndrome, bilateral adrenalectomy is recommended. Hormone replacement is required in this setting. Although not curative, debulking surgery for control of symptoms is the primary therapy for malignant pheochromocytoma.
The radioisotope iodine-131 metaiodobenzylguanidine (131I-MIBG) has been used as a therapeutic agent in malignant pheochromocytomas that demonstrate avid uptake of the agent. Investigators have reported partial responses, based on biochemical response as well as decreased tumor volume, ranging from 18% to 82%.33–35 Symptomatic improvement was observed in responding patients with regard to both painful metastases and manifestations of increased catecholamine levels. Partial remissions are usually temporary, with some patients relapsing between doses of MIBG. In other patients, sustained partial remissions have been noted with durable palliation extending 2 to 3 years.34,36 Prolonged survival has been associated with measurable responses and higher administered doses of MIBG (>500 mCi).37 Toxicity includes bone marrow toxicity (particularly thrombocytopenia), nausea, and vomiting.
Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine has shown efficacy in a small study. In 14 treated patients, the clinical and biochemical response rates were 57% and 79%, respectively. Response was associated with objective improvement in performance status and blood pressure, and treatment was well tolerated.38
 RADIATION THERAPY TECHNIQUES
RADIATION THERAPY TECHNIQUES
The role of radiation in the management of ACC is controversial. Locoregional disease control remains a major problem in this disease, and some reports suggest that external radiation may reduce recurrence rates.
In the primary management of ACC, external radiation may play a role either preoperatively for unresectable tumors, postoperatively for patients with residual disease or high risk of local failure, or as definitive therapy for patients who are medically unfit for surgery. Radiation is also effective in the palliative setting for bone and nodal metastases.
For patients with macroscopic or unresectable disease, doses of 50 to 60 Gy delivered during 5 to 6 weeks should be considered. Initial fields should encompass the gross tumor with adequate margins as well as the regional lymph nodes, which should include the contralateral para-aortic lymph nodes. Dose to regional nodes can be limited to 45 Gy when they are not macroscopically involved. Care should be taken to limit dose to the spinal cord, kidneys, liver, and small bowel. For macroscopic disease for which high dose is desired, conformal techniques and intensity-modulated radiation should be considered. For patients receiving postoperative treatment for high-risk or microscopic disease, doses of 45 to 54 Gy are appropriate.
In the palliative setting, doses of 30 to 40 Gy given during the course of 2 to 3 weeks are reasonable. In patients with painful bone metastases, hypofractionated regimens should be considered for patients with poor performance status or otherwise limited life expectancy.
External-beam radiation is limited to a palliative role in the management of pheochromocytomas.
 FOLLOW-UP
FOLLOW-UP
Patients with pheochromocytomas should have frequent follow-up that includes a thorough history and physical examination with an evaluation of vital signs and plasma markers. This should be done every 6 months for the first 3 years starting around 3 months postsurgery and annually thereafter. Advanced or persistent disease may require more frequent follow-up and symptom management.
 REFERENCES
REFERENCES
1. Moore K, Agur A. Essential clinical anatomy. Baltimore: Lippincott Williams & Wilkins, 2002:182–185.
2. Rouvière H. Anatomie des lymphatiques de l’homme. Paris: Masson, 1932.
3. Dunnick NR. Adrenal carcinoma. Radiol Clin North Am 1994;32:99–108.
4. McClennan BL. Oncologic imaging. Staging and follow-up of renal and adrenal carcinoma. Cancer 1991;67:1199–1208.
5. Shambaugh E, Ryan R. Summary staging guide for the cancer surveillance, epidemiology and end results reporting (SEER) program. Rockville, MD: US Dept of Health and Human Services, Public Health Service, 1977.
6. Berruti A, Terzolo M, Sperone P, et al. Etoposide, doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr Relat Cancer 2005;12:657–666.
7. Bravo EL. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev 1994;15:356–368.
8. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int 1991;40:544–556.
9. Cotran A, Kumar V, Robbins SL. Robbins pathologic basis of disease, 5th ed. Philadelphia: WB Saunders, 1994:1161–1164.
10. Hubbard MM, Husami TW, Abumrad NN. Nonfunctioning adrenal tumors. Dilemmas in management. Am Surg 1989;55:516–522.
11. De Maria M, Barbiera F, Bonadonna F, et al. Diseases of the adrenal medulla. Rays 1992;17:62–86.
12. Miller RW, Fraumeni JF Jr, Hill JA. Neuroblastoma: epidemiologic approach to its origin. Am J Dis Child 1968;115:253–261.
13. Beard CM, Sheps SG, Kurland LT, et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc 1983;58:802–804.
14. Melicow MM. One hundred cases of pheochromocytoma (107 tumors) at the Columbia-Presbyterian Medical Center, 1926–1976: a clinicopathological analysis. Cancer 1987;40:1987–2004.
15. Beierwaltes WH, Sisson JC, Shapiro B. Malignant potential of pheochromocytoma. Proc Am Acad Cancer Res 1986;27:617.
16. Cryer PE. Phaeochromocytoma. Clin Endocrinol Metab 1985;14:203–220.
17. Javadpour N, Woltering EA, Brennan MF. Adrenal neoplasms. Curr Probl Surg 1980;17:1–52.
18. Percarpio B, Knowlton AH. Radiation therapy of adrenal cortical carcinoma. Acta Radiol Ther Phys Biol 1976;15:288–292.
19. Loughlin KR, Gittes RF. Urological management of patients with von Hippel-Lindau’s disease. J Urol 1986;136:789–791.
20. Nakagawara A, Ikeda K, Tsuneyoshi M, et al. Malignant pheochromocytoma with ganglioneuroblastoma elements in a patient with von Recklinghausen’s disease. Cancer 1985;55:2794–2798.
21. Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer 1993;72:3145–3155.
22. Haak HR, Hermans J, van de Velde CJ, et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer 1994;69:947–951.
23. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution’s experience. Medicine 1991;70:46–66.
24. King DR, Lack EE. Adrenal cortical carcinoma: a clinical and pathologic study of 49 cases. Cancer 1979;44:239–244.
25. Magee BJ, Gattamaneni HR, Pearson D. Adrenal cortical carcinoma: survival after radiotherapy. Clin Radiol 1987;38:587–588.
26. Markoe AM, Serber W, Micaily B, et al. Radiation therapy for adjunctive treatment of adrenal cortical carcinoma. Am J Clin Oncol 1991;14:170–174.
27. Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery 1992;112:963–971.
28. Terzolo M, Angeli A, Fassnacht M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med 2007;356:2372–2380.
29. Ahlman H, Khorram-Manesh A, Jansson S, et al. Cytotoxic treatment of adrenocortical carcinoma. World J Surg 2001;25:927–933.
30. Lo CY, Lam KY, Wat MS, et al. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg 2000;179:212–215.
31. Plouin PF, Duclos JM, Soppelsa F, et al. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocr Metab 2001;86:1480–1486.
32. Amar L, Servais A, Gimenez-Roqueplo AP, et al. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocr Metab 2005;90:2110–2116.
33. Shapiro B, Gross MD, Shulkin B. Radioisotope diagnosis and therapy of malignant pheochromocytoma. Trends Endocrinol Metab 2001;12:469–475.
34. Shapiro B, Sisson JC, Wieland DM, et al. Radiopharmaceutical therapy of malignant pheochromocytoma with [131I]metaiodobenzylguanidine: results from ten years of experience. J Nucl Biol Med 1991;35:269–276.
35. Sidhu S, Sywak M, Robinson B, et al. Adrenocortical cancer: recent clinical and molecular advances. Curr Opin Oncol 2004;16:13–18.
36. Loh KC, Fitzgerald PA, Matthay KK, et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest 1997;20:648–658.
37. Safford SD, Coleman RE, Gockerman JP, et al. Iodine-131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery 2003;134:956–962.
38. Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med 1988;109:267–273.
< div class='tao-gold-member'>




