Donor Lymphocyte Infusions Following Stem Cell Transplantation
Investigators have attempted to treat posttransplant viral diseases and relapsed malignancies by infusion of donor lymphocytes (DLI) since this population contains virus-reactive and potentially tumor-specific T cells.
Donor Lymphocytes to Treat Viral Infections. Serious viral infections contribute significantly to morbidity and mortality following HSCT, and the most frequent of these include CMV, EBV, AdV, BK polyoma virus (BKV), human herpes virus 6 (HHV6), respiratory syncytial virus (RSV), and metapneumovirus (MPV). While pharmacological therapies such as ganciclovir (GCV) or foscarnet are effective treatment for CMV, many other viruses lack established therapies. Even for CMV, treatment may be associated with toxicities such as myelosuppression or renal dysfunction and may lead to the development of drug-resistant viral strains. Under physiological conditions, T cells play a critical role in control of these viral infections, and it is the diminished number and function of these cells after HSCT that contributes to the increased incidence of viral disease. Indeed, recipients who receive ex vivo T-cell depleted stem cell products or T-cell depleting antibodies as part of their conditioning regimen have a particularly high morbidity and mortality from these disorder. Although DLIs have successfully been employed to treat EBV, CMV, and AdV infections post-HSCT,
66,67,68 their use is limited by the presence of alloreactive T cells with the DLI product that can cause graft-versus-host disease (GVHD), a potentially life-threatening complication. Several strategies have been developed to reduce the risk of GVHD from DLI, either by depleting alloreactive T cells, genetically modifying T cells with “suicide genes,” or enriching cell products for virus-specific T cells (see section “
Virus-specific T cells”).
Donor Lymphocytes to Treat Malignancies. Adoptive immunotherapy with DLI after HSCT effectively augments the graft-versus-leukemia response and can eliminate residual disease, especially in patients with chronic myelogenous leukemia (CML).
69,70 In the original studies, three patients with relapsed CML attained complete cytogenetic remissions after treatment with DLI and IFNα post-HSCT. In larger series, approximately 70% of all relapsed CML patients treated in chronic phase achieved complete cytogenetic remission but in accelerated phase or blast crisis just 11% had a CR. DLI are less effective treatment for patients who relapse with other hematological malignancies after HSCT, with a response rate of 29% for AML and 5% for ALL.
69Although this DLI-mediated graft-versus-leukemia (GVL) effect may simply be another manifestation of GHVD, recent studies indicate that minor histocompatibility antigen (mHAgs)-specific
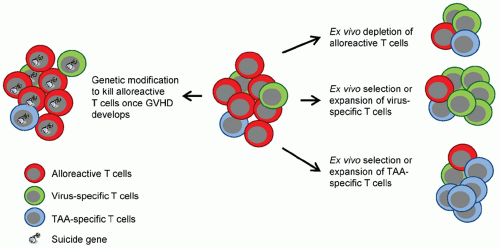
T cells present in DLIs can induce durable remissions of malignancies without causing GVHD. Nonetheless, the application of DLI remains limited by the frequency with which it induces GVHD. Thus, investigators have attempted to ameliorate GVHD while sparing GVL by infusing allodepleted and suicide gene-modified donor lymphocytes. In addition, investigators have isolated donor-derived T cells that recognize virus-specific T cells (see section “
Virus-specific T cells”) or TAA-specific T cells as discussed in section “Donor-derived, antigen-specific T cells” (
Fig. 14.3).
Allodepleted and Suicide Gene-modified Donor Lymphocytes. Infusing CD8-depleted lymphocytes reduces the incidence of GVHD with preservation of “graft versus tumor” effects.
71 However, due to varying degrees of alloreactivity with different donor-recipient pairs, it has proven difficult to define a T-cell dose that has antitumor activity but does not cause GVHD. Several approaches have therefore been developed to reduce the risk of GVHD or to specifically eliminate infused T cells once GVHD develops.
One means of overcoming the problem of alloreactivity is to remove alloreactive cells from the T-cell product on the basis of these cells’ upregulation of activation markers in response to stimulation by alloantigens. Several studies have evaluated this strategy using an immunotoxin directed against the activation marker CD25. This procedure enables the in vitro depletion of alloreactive cells while preserving T cells reactive to viruses and tumor-associated antigens such as the minor histocompatibility antigen HA1 and primary granule enzyme proteinase 3. In two Phase I clinical studies, patients received T cells depleted of alloreactive cells, and early T-cell expansion was seen with improvements in immune reconstitution against viral pathogens without significant GVHD.
72,73 Besides CD25 immunotoxin, a photoactive rhodamine derivate, TH9402, has also been used to deplete alloreactive T cells ex vivo prior to T-cell infusion.
74An alternative approach relies on the introduction of a suicide gene into T cells so that cell death can be induced in vivo once GVHD develops. For example, donor-derived T cells transduced with a retroviral vector expressing human herpes simplex virus thymidine kinase (HSV-
tk), which renders cells sensitive to GCV, have been infused into HSCT recipients. These modified T cells were safe and accelerated immune reconstitution, and any GVHD was successfully controlled with GCV.
75 The limitations of this approach are the inherent immunogenicity of HSV-
tk, which may lead to unwanted elimination of the modified cells, and the requirement of nucleoside analogs such as GCV for T-cell killing, which precludes their use as antiviral agents. As an alternative, killing may be induced by introducing dimerizable death molecules such as inducible caspase 9 (iC9) that can be triggered by a small-molecule dimerizing drug or by using monoclonal antibodies directed to cell surface antigens, such as CD20 (using rituximab) or EGFR (using cetuximab) introduced into the T cells.
76,77,78,79 iC9 gene-modified, allodepleted donor-derived T cells were evaluated in haploidentical HSCT recipients.
76 A single dose of a small-molecule drug (AP1903) that dimerizes and activates the iC9 transgene eliminated more than 90% of iC9 gene-modified T cells within 30 minutes of administration and eliminated GVHD without recurrence. Long-term follow-up of these patients revealed accelerating immune reconstitution, resulting in protection against major viral pathogens including EBV, CMV, AdV, and BKV.
80
Polyclonal-activated T Cells
T cells in cancer patients are often anergic, that is, unresponsive when they are stimulated through their T-cell receptor. One potential strategy to reverse anergy is to nonspecifically activate T cells ex vivo outside the immunosuppressive environment of the cancer patient prior to reinfusion. Ex vivo activation of T cells has been most extensively evaluated using CD3-antibody-stimulated T cells. Most clinical studies have been in patients with metastatic renal cell carcinoma, but initially encouraging results have not been replicated in larger studies.
81 Similarly, a study using autologous activated T cells as adjuvant therapy after complete resection of hepatocellular carcinoma showed no influence on overall survival, although infused patients had significantly longer recurrence-free survival.
82 Activation of T cells with anti-CD3- and anti-CD28-coated beads ex vivo may have greater potential to overcome disease-induced anergy and augment CD4-positive T-cell responses. Several clinical studies have been conducted with anti-CD3/anti-CD28 autologous, activated T cells,
83,84,85,86 in which the patients received activated T cells after autologous HSCT. T-cell infusions induced a rapid recovery of lymphocyte counts and reversed cytokine activation deficits in vitro. In a subset of patients, T-cell infusions were associated with a clinical picture indistinguishable from acute GVHD.
84In one study, donor-derived, activated T cells were given to patients after allogeneic HSCT. Infusions were safe and not associated with an increased risk of GVHD. The impact on disease and relapse was unclear, and further studies will be needed to determine if this approach is superior to DLIs. In an effort to increase the frequency of antigen-specific T cells in autologous, activated T-cell
products, multiple myeloma patients have been vaccinated with an influenza virus vaccine, pneumococcal vaccine, and/or a peptide vaccine targeting the TAAs survivin and telomerase prior to leukapheresis and ex vivo T-cell activation. This resulted in improved recovery of influenza virus-specific or pneumococcal-specific immune responses post-infusion in comparison with patients, who did not receive the vaccine.
83,86,87 While approximately one-third of patients who received the survivin and telomerase peptide vaccine developed survivin- and/or telomerase-specific immune responses, the responses were in general significantly lower when compared with the coadministered pneumococcal vaccine, most likely explaining why the induction of surviving- and/or telomerase-specific immune responses did increase event-free survival rate in comparison with nonresponders.
87
Tumor-infiltrating Lymphocytes
TILs can be isolated from 30% to 65% of human solid tumors and are usually CD4-positive T cells except in melanoma, in which CD8-positive T cells predominate.
88 As the number of TILs isolated from tumor biopsies is insufficient for adoptive immunotherapy protocols, they must be expanded ex vivo prior to infusion, using cytokines such as IL-2. Expanded CD8-positive TIL have cytolytic activity against the original tumor and, unlike LAK cells, their killing is MHC class I restricted.
3 The varied success of clinical trials using TIL likely reflects differences in tumor immunogenicity and tumor burden at the time of TIL infusion.
89 To increase the antitumor efficacy of TILs, patients have been lymphodepeleted with fludarabine (Flu) and cyclophosphamide (Cy) with 0, 2, or 12 Gy of total body irradiation (TBI) prior to TIL transfer.
7,90 Lymphodepletion in 93 patients with metastatic, refractory melanoma increased the levels of IL-7 and IL-15 in peripheral blood and increased the objective response rate from 49% in patients receiving only Flu/Cy to 72% in patients receiving Flu/Cy and 12 Gy TBI. Telomere length and the expression of cell surface markers such as CD27 have been correlated with antitumor activity of infused TILs.
91The specificity of T cells in TILs is actively being investigated and studies have identified mutated antigens within tumors, such as kinesin family member 2C (KIF2C)-mutated DNA polymerase alpha subunit B (POLA2) or erbb2 interacting protein (ERBB2IP) as TIL targets.
92,93 While the majority of clinical responses have been observed in melanoma patients receiving TILs enriched in CD8-positive T cells, a recent case report highlights the antitumor activity of CD4-positive TILs in a patient with metastatic cholangiocarcinoma.
93 Overall, the variable success of isolation and expansion of TILs from tumor samples other than melanoma means that adoptive transfer of TILs is not currently a viable option for pediatric malignancies.
94