Adduct-Forming Agents: Alkylating Agents and Platinum Analogs
Since the first clinical experiments with nitrogen mustard at Yale in the early 1940s, alkylating agents have played a primary role in cancer treatment (1). The early mustards have gradually been replaced by platinum-based compounds in most regimens for treating epithelial cancers, but remain primary components in the treatment of childhood solid tumors, lymphomas, and adult sarcomas, and in high-dose chemotherapy. As a class, they have the features of the prototypical cytotoxic drugs, with broad antitumor activity, but they adversely affect many normal tissues as well. They share common characteristics of a significant increase in response as doses are escalated: acute toxic effects on bone marrow, epithelium of the gastrointestinal tract, and hair follicles; significant toxicity to lung, heart, and central nervous systems at bone marrow ablative doses; and late induction of myelodysplasia and acute leukemia.
MECHANISM OF ACTION
Three general classes of DNA adduct-forming agents have found clinical application. The first are the chloroethyl nitrogen mustards, exemplified by cyclophosphamide, ifosfamide, melphalan, and chlorambucil (Figure 5-1). These drugs become active through formation of a highly reactive imonium intermediate, which transfers its ethyl group to nucleophilic (electronegative) sites on DNA (amino, hydroxyl, or phosphate sites) and to sulfhydrils on amino acids and glutathione (Figure 5-2). These drugs contain two chloroethyl groups and can cross-link DNA, creating a lesion that is difficult to repair. The classical nitrogen mustard was a highly unstable molecule, while the current agents of this type, such as cyclophosphamide and melphalan, have been modified by conjugation with electronegative groups that reduce their reactivity. The single strand adducts are repaired by nucleotide excision repair, while double strand breaks require the complex homologous recombination system. The newest agent of this class, bendamustine (Figure 5-1), consists of a purine-like ring, to which is attached a classical bifunctional nitrogen mustard. This bulky molecule forms adducts that are more slowly repaired. The drug is incompletely cross-resistant with traditional alkylators and is highly effective for chronic lymphocytic leukemia and follicular lymphomas (2).
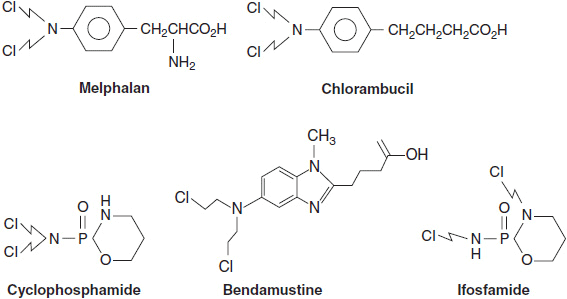
FIGURE 5-1 Molecular structures of melphalan, chlorambucil, cyclophosphamide, ifosfamide, and bendamustine.
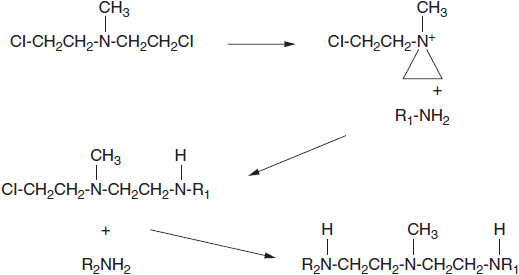
FIGURE 5-2 Nitrogen mustard undergoes spontaneous chemical rearrangement in aqueous solution to form a highly reactive, positively charged, three-member aziridinium ring, which reacts with nucleophilic sites on DNA such as amines and hydroxyl groups. (R1NH2 and R2NH2 represent reactive sites on DNA bases.)
The second group consists of drugs that transfer single methyl radicals to DNA. This second class—which includes procarbazine, dimethyl triazinoimidazolecarboxamide (DTIC); its close congener, temozolomide; and busulfan—requires more complex activation, either enzymatic or chemical. The methylating drugs preferentially attack the O-6 position of guanine, as well as other reactive sites on purines and pyrimidines. The methylation of O-6 guanine is repaired by methyl guanine-O-6 methyltransferase, (MGMT). The activity of MGMT in cancer cells determines the response of primary brain tumors to this group of drugs.
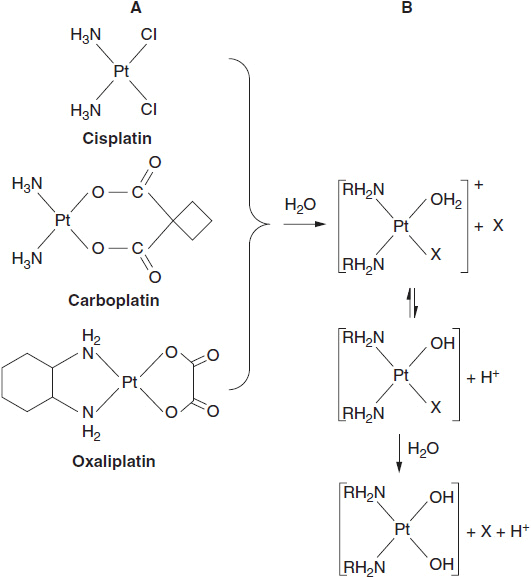
The third group of drugs, platinum complexes, closely mimic alkylators, as they form reactive intermediates that attack the same nucleophilic sites on DNA. The activation of these analogs begins with the displacement of chloride or other leaving groups by water, with the formation of a reactive hydroxyl intermediates (Figure 5-3). The platinum analogs have two leaving groups, either chlorides or an oxalate group, and thus are capable of cross linking DNA. Their adducts are repaired in a manner similar to the chloroethyl alkylators.
 CELLULAR PHARMACOLOGY OF ALKYLATING AGENTS
CELLULAR PHARMACOLOGY OF ALKYLATING AGENTS
As a class, most alkylating agents are lipid soluble and easily cross cell membranes. Two members of the class, nitrogen mustard and melphalan are actively transported into cells, the former by the choline transporter and the latter by amino acid transporters. Resistance may arise experimentally by deletion of these transporters.
Following uptake, most alkylating agents, and in particular the chloroethyl mustards, spontaneously undergo activation to an unstable, electrophilic intermediate. Some agents, such as DTIC, require metabolic activation, but the closely related temozolomide generates the same unstable methyltriazino derivative spontaneously, and has become the standard agent for treating gliomas. Cyclophosphamide and ifosfamide require prior p450 oxidative activation in the liver in order to generate their chemically reactive end products, which are phosphoramide mustards (Figure 5-4).
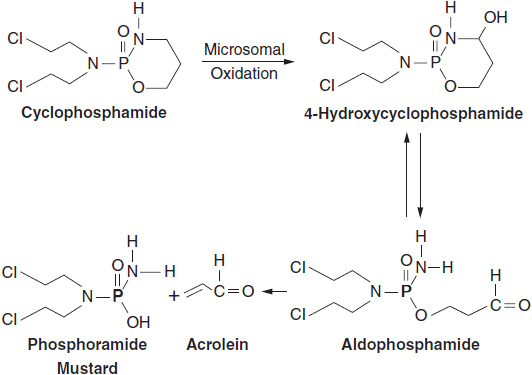
Once inside cells, chloroethyl alkylating agents form reactive imonium ions (Figure 5-2), which attack sites such as sulfhydryl groups on proteins and glutathione (the sulfhydryl found in highest concentration within cells). They also react with electron-rich sites on nucleic acids, including the N-2 and N-7 groups on guanine, the O-6 on guanine, and the N-3 on adenine. Single-base alkylations are recognized by the DNA nucleotide excision repair (NER) complex, while cross-links must be repaired by a more complex process of excision of the alkylated bases and adjoining segments, and accurate repair of the resulting double stand break through homologous recombination. An accumulation of cross-links and strand breaks, particularly interstrand breaks, signals p53 to initiate apoptosis (3).
Specific enzymes may be critical to the process of DNA repair and survival. Methylating agents and the nitrosoureas have a particular propensity for alkylation of the O-6 position of guanine, and this alkylation is removed by MGMT. The MGMT gene is methylated and silenced in 20% of primary brain tumors, and these tumors have greater sensitivity and a better prognosis when treated with procarbazine, temozolomide, or the nitrosoureas.
The ability to repair alkylating damage is likely an important determinant of antitumor response. Repair capacity varies among tumors. This variation may result from inherited polymorphisms in NER components, notably ERCC1. Low levels of expression of ERCC1 have correlated with greater response in lung, ovarian, and head and neck cancers (4), but results are inconsistent in other studies. Polymorphisms of enzymes in the NER pathway have also been correlated with greater host toxicity in lung cancer patients (5). Inherited or somatic changes in the double strand break repair pathway may also influence response. BRCA1- or BRCA2-deficient cells have impaired double strand break repair and exhibit heightened sensitivity to platinum analogs (6), a finding confirmed in some breast cancer trials with triple negative tumors (7).
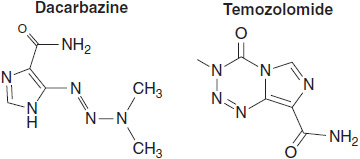
FIGURE 5-5 Dacarbazine, Temozolomide
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


