An increasing number of inherited clinical conditions with associated genomic changes have been defined that predispose individuals to develop MDS/AML. In such individuals, the development of AML often emerges after a period of MDS. In these cases, such as Fanconi anemia or DS, both the patient and the AML may have distinctive characteristics requiring alternative approaches to therapy. In other situations, such as in germline mutations of CEBPA or RUNX1, the AML more closely resembles the de novo AML that arises in other children and is similarly treated.
Constitutional Chromosomal Abnormalities and MDS/AML
The occurrence of multiple cases of leukemia in some families has been known for decades. Such families usually have more than one first-degree relative affected with AML and/or MDS, which tends to occur early in life.
Constitutional chromosomal abnormalities leading to MDS/AML predisposition have been reported from Japan in approximately 13% of pediatric patients with MDS.
31 In a population-based study from British Columbia, patients with known predisposing conditions represented 48% of the cases of MDS, but 23% of the total was represented by children with trisomy 21 (DS).
32With refinement in karyotypic analysis, it became clear that some of this familial predisposition was due to constitutional, chromosomal abnormalities, such as translocations of t(7;20) and t(3;6), as well as inheritance of monosomy 7.
33,34,35 Estimates have been reported that up to 10% of familial MDS is associated with monosomy 7 and sometimes del(7q).
32,36 Familial monosomy 7 may also be associated with neurologic problems, such as cerebellar ataxia.
37 However, the question has been raised that another chromosomal locus may be a key driver in the development of MDS in these families in part because of the relatively low incidence of leukemia in individuals with chromosome 7 constitutional abnormalities.
38 Additional evidence has come from the observation of the different parental origin of the remaining chromosome 7 in siblings with monosomy 7.
33Constitutional trisomy 8, including the presence of mosaicism, has been reported to be associated with MDS and AML in 15% to 20% of cases.
39 There are also reports of MDS and AML in patients with Klinefelter (47, XXY) and Turner (45, X) syndromes.
40,41,42 However, increased risk for these two syndromes has not been documented in larger cohort studies.
43 Whether there is an association of MDS/AML in children who are born with congenital anomalies unrelated to other known causes is unclear. A study from the COG demonstrated that the presence of trisomy 21 (DS) was the primary risk factor for the development of leukemia and not an association with the presence of congenital anomalies alone.
44Down syndrome represents the most common inherited condition that has the highest incidence of leukemia with a 10- to 20-fold associated risk compared with those without DS.
45 Of note, a population-based study has reported up to a 50-fold risk under 5 years of age but with 150-fold for AML and 40-fold for ALL. This is in contrast to the relatively equivalent incidence of AML and ALL in children with DS later in life.
46,47 Of note, the risk of leukemia in individuals with DS decreases to about 10-fold up to age 29, and then after 30 years of age, the risk is similar to that of individuals without DS.
47 Overall, the cumulative risk of patients with DS developing leukemia before 5 years of age is about 2%, and by the age of 30 years is 2.7%.
47The most common subtype of leukemia in this group of patients during the first 3 years of life is acute megakaryoblastic leukemia (AMKL), as defined by morphological and immunophenotyping criteria.
46 DS is also associated with another distinctive myeloid disorder, called “transient myeloproliferative disorder (TMD)” or “transient abnormal myelopoiesis (TAM).” TMD occurs in approximately 10% of neonates with DS and characteristically mimics the presentation of AML. Although TMD usually spontaneously remits over the course of weeks to a few months, approximately 20% to 30% of cases of TMD develop AMKL by the age of 4 years.
48,49,50 The high percentage of patients who have spontaneous remission of TMD led to the alternative terminology of “transient leukemia.” Importantly, some children may not have the typical clinical characteristic of DS due to trisomy 21 mosaicism. These children also have increased risk of developing TMD and AML that arise from the hematopoietic precursors with the trisomy 21.
48,49,50Mutations of the
GATA1 transcription factor gene are nearly always found in DS-associated TMD, while additional mutations may be required for the development of AML.
51,52,53,54 One study reported that no
GATA1 mutations were observed in fetuses with DS up to 12 to 25 weeks of gestation, strongly suggesting that they occur relatively late during development.
55 Additional secondary mutations reported in AMKL associated with DS have included cohesion genes,
CTCR, EZH, KaNSL1, and other epigenetic regulators plus
JAK, MPL, SH2B3, and
RAS pathway-associated genes. Of note, in this report, only mutations of
GATA1 were observed in TMD cases.
56 In another report, mutations in other genes were seen in the AMKL arising from TMD but also in two TMD cases in which GATA1 mutations were not identified. In another case of TMD, two different
GATA1 mutations were identified, but the AMKL that developed arose from only one of the two mutated
GATA1 clones (
Fig. 20.3).
57
Inherited Cytopenias of Selective Lineages
Severe congenital neutropenia (SCN), also called Kostmann syndrome, represents an inherited granulocytopenia with a 25% estimated 10-year cumulative risk of MDS/AML and with a long-term annual risk of 2.3% per year after 10 years.
79,80 Children present during infancy with absolute neutrophil counts of less than 500/µL and have significant bacterial infections.
SCN is primarily caused by inherited mutations in neutrophil elastase (
ELANE) gene (previously termed
ELA2). However, there are cases of SCN that do not have mutations of
ELA2, but still have an increased risk of developing MDS/AML (
Fig. 20.4). Mutations of the
HAX1 gene, which encodes a heat shock associate protein-linked LYN, a key SRC family tyrosine kinase that plays a key role in myelopoiesis, have been described in SCN. Neurological abnormalities are associated with patients with SCN due to
HAX1 gene mutations.
81 The mechanism(s) leading to neutropenia is unclear for these gene defects, but likely linked to apoptosis resulting from misfolded elastase proteins or to a role of HAX1 in mitochondria-regulated apoptosis.
82Further, as Next-Gen sequencing approaches to such disorders have increasingly been used, other mutations have been identified, such as those involving
CXCR2, which encodes a chemokine receptor that in turn transduces interleukin 8 (IL8)-mediated signals through a G-protein-activated signal transduction cascade.
83 Mutations of the
VPS45 gene, which encodes a protein involved in endosomal membrane trafficking, have been reported in familial neutropenia, neutrophil dysfunction, bone marrow fibrosis, and nephromegaly.
84 Mutations involving the
ELANE gene, some of which overlap with those associated with SCN, have also been identified in cases of cyclic neutropenia, which, however, is not associated with an increased incidence of MDS/AML.
85,86,87 Mutations in the glucose 6-phosphate catalytic subunit 3 (
G6PC3) gene and the transcription factor encoding
GFI1 gene have also been linked to cases of SCN.
88 Mutations affecting the Wiskott-Aldrich syndrome gene (
WAS) have also been linked to cases of SCN.
88The bone marrow of such patients reveals a decreased production of granulocytic lineage and usually a maturational arrest before or at the promyelocytic/myelocytic stage. In addition,
neutrophils that are produced in SCN have been reported to have abnormal granule maturation and decreased antimicrobial activity. The increased production of neutrophils associated with recombinant G-CSF exposure does not appear to abrogate some of these maturational, functional defects.
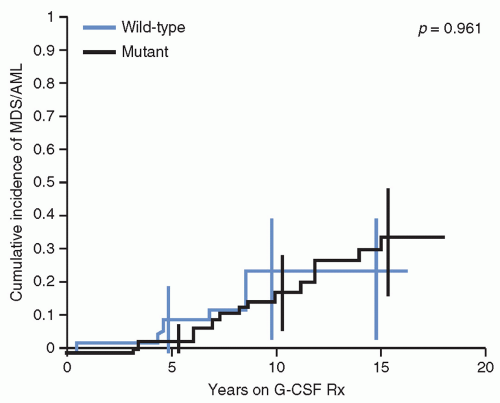
89 Nevertheless, patients with SCN usually benefit from treatment with recombinant G-CSF, which increases neutrophil counts and reduces the risk of fatal infections. However, the use of G-CSF has been associated with an increased frequency of MDS/AML, estimated to be about 2.3% per year, in a dose-dependent manner.
79,90 Of note, the risk of leukemia appears to be similar in patients with or without
ELANE mutations (
Fig. 20.4).
79,80,91G-CSF receptor (
CSF3R) mutations have been identified in approximately 80% of cases of SCN and AML, strongly suggesting that these mutations contribute to the development of AML as a second genetic hit.
92 In addition, coexistence of cooperating
CSF3R and
RUNX1 mutations has been reported in SCN.
93Thus, patients with SCN are usually clinically followed with both complete blood counts on a regular basis as well as with yearly bone marrow examinations to detect morphologic, chromosomal, and gene mutations associated with the evolution of MDS/AML. In such cases, early hematopoietic stem cell transplantation (HSCT) before progression to AML can be both curative of the SCN and prevent emergence of leukemia. It has been recommended that patients with SCN and a poor response to G-CSF should be referred for allogeneic HSCT during the first year of life.
94,95,96,97,98Neurofibromatosis Type 1 (NF1), along with Noonan, Costello, LEOPARD, and SPRED1 syndromes, all represent disorders considered part of the inherited Neuro-Cardio-Facio-Cutaneous (NCFC) disease family. They all are the result of inherited gene alterations that lead to activation of the RAS-BRAF-MAP-ERK pathway critical in hematopoietic cell proliferation and survival. These syndromes are also cancer predisposition syndromes that are associated with both MDS/AML (particularly LEOPARD, Costello, and SPRED1 syndromes) and with JMML (NF1 and Noonan Syndrome).
99,100,101,102,103
CEBPA-mediated familial AML is a rare disorder with autosomal dominant inheritance of germ line mutations involving the
CEBPA, which encodes a CCAAT/enhancer-binding transcription factor and master regulator of myelopoiesis.
104 Somatic mutations of
CEBPA are also commonly observed in sporadic cases of AML. The AML that arises as a result of
CEBPA mutation is often characterized as differentiated with the presence of Auer rods and expression of the lymphoid antigen, CD7.
105
Germ line mutations in the key hematopoietic transcription factor,
GATA2, have also been reported in patients with familial
CEBPA mutations, but not in those without
CEBPA mutations, suggesting the need for cooperation for the development of AML.
106 Germ line mutations of
GATA2 have also been reported in patients with MDS/AML in conjunction with profound circulating monocytopenia, B- and NK-cell lymphopenia, pulmonary alveolar proteinosis, and susceptibility to multiple opportunistic infections. This autosomal dominant inherited syndrome, termed the “Mono-MAC” syndrome, has a median onset of 32 years of age, but has been reported in children as young as 7 years.
83,107Mutations of these genes in MDS/AML should alert clinicians to consider that such patients may have such inherited predisposition conditions.
108Shwachman-Diamond syndrome (SDS) is an autosomal recessive disorder characterized by exocrine pancreatic insufficiency, heart and skeletal abnormalities (e.g., short stature, osteopenia, metaphyseal dysostosis), and neurocognitive problems and BMF, particularly neutropenia, with a significant predisposition for MDS/AML.
109 With a median follow-up of 11.6 years, a French national cohort study of 102 patients with SDS showed that there was a 20-year cumulative risk of severe cytopenia of 24.3%; first symptoms appearing before 3 years of age and significantly low peripheral blood counts were associated with the most severe hematologic complications.
110 Out of a total of 102 patients, 12 patients developed what were considered malignant cytopenias; if only the 41 patients who had severe cytopenias were considered, then about 29% developed MDS/AML (
Fig. 20.5).
110 An NCI study from the United States observed AML in patients with FA and DC, but none in 17 patients with SDS.
65 The difference is likely due to the smaller number of patients examined in this study compared with the National French Cohort. The evolution to MDS/AML in patients with SDS is also associated with characteristic accompanying chromosomal abnormalities, including those involving chromosome 20 and isochromosome 7q. The del20(q11) involves a region known to be involved in patients with MDS/AML without SDS. The isochromosome 7q defect cases more often had stable persistence of the abnormal clone that may be dependent on
SBDS gene dosage variability located in this chromosomal region.
111
SDS is caused in about 90% of diagnosed patients by mutations in the
SBSD gene, which encodes for a critical cofactor associated with Elongation Factor 1 (ELF1). Together they catalyze the GTP-dependent release of EIF6 from the nascent 60S ribosomal subunit, allowing it to effectively combine with the 40S ribosomal subunit in order to effect protein synthesis. This defect in ribosomal biosynthesis results in altered mRNA utilization and protein synthesis, which in turn stimulates a p53 response and apoptosis.
109,112Diamond-Blackfan anemia (DBA) is another important BMF syndrome characterized by skeletal anomalies (triphalangeal thumbs, short stature), craniofacial defects (hypertelorism, flat nasal bridge, cleft palate), cardiac abnormalities, red blood cell aplasia, and an increased risk of malignancy.
113,114,115 DBA is also another ribosomopathy in that the majority of the etiologically linked mutated genes encode for proteins that comprise the large and small ribosome subunits. While mutated ribosomal protein-encoding genes represent approximately 25% of patients with
DBA, approximately another 25% of patients may have segmental gene or chromosomal deletions involving ribosomal protein-encoding genes.
116,117 The ribosomal protein-encoding gene,
RPS14, has also been shown to cause the 5q2 MDS syndrome that can mimic DBA, but responds to lenalidomide therapy.
118,119,120 Such defects in ribosome biosynthesis in DBA have been shown to result in increased TP53 activity and apoptosis, although it is unclear why such a defect results primarily in erythroid lineage aplasia in DBA and primarily granulocytic decreases in SDS. Mutations of the
GATA1 transcription factor have also been reported to result in a DBA phenotype.
121,122,123 These observations have led to the proposal that GATA1 defects result in altered expression of ribosomal protein genes, thus resulting in a similar phenotype to a primary ribosomal protein-encoding gene defect.
122
How such ribosome haploinsufficiency syndromes lead to cancer, and particularly MDS/AML, is unclear. However, the observed to expected ratios of MDS and AML in patients with DBA have been reported as 287 and 28, respectively.
124 Therefore, these patients need to be monitored clinically for management of their anemia but also for the emergence of MDS/AML and other neoplasias.
The familial platelet disorder (FPD) with the propensity to develop AML is a result of germ line mutations in the
RUNX1 (previously termed
CBFA2,
AML, and
PEBP2AB) gene that encodes for the Runt-related transcription factor 1.
125,126,127 Patients usually present in infancy with mild-to-moderate thrombocytopenia and bleeding tendency, but without dysmorphia.
125 Some cases with associated mental retardation, dysmorphic features, and short stature have been described and appear to be related to large deletions that involve
RUNX1 and other genes.
128 Germ line mutations of
RUNX1 have also been associated with T ALL.
129 A syndrome of congenital transfusion-dependent thrombocytopenia and anemia with MDS has been linked to germ line mutation of
GATA1.
130
Congenital amegakaryocytic thrombocytopenia (CAMT) usually presents at birth without any physical abnormalities but with isolated thrombocytopenia with decreased bone marrow megakaryocytes; it then progresses to BMF during the first few years of life. The disorder has been shown to be due to either homozygous or compound heterozygous mutations of the
c-MPL gene, which encodes for the thrombopoietic receptor.
131,132,133,134 The lifelong risk of developing MDS/AML has been estimated to be less than 10%. Patients usually undergo HSCT, which is curative. The molecular basis for this thrombopoietic receptor defect leading to trilineage BMF remains unclear, but may relate to the type of mutation affecting the receptor and its role in earlier hematopoiesis.
135
Thrombocytopenia with absent radius (TAR) syndrome has also been associated through case reports with the development of MDS/AML. The syndrome is linked to deletions involving chromosome 1q21.1, and is associated with several low-level regulatory single nucleotide polymorphisms (SNPs) as well as a null mutation in the
RBM8A gene, which is an RNA-binding protein involved in RNA splicing.
136 Hematopoietic precursors have been reported to have defective thrombopoietin signaling, particularly in the fetal and neonatal period, so that the thrombocytopenia usually resolves within the first year of life.
137
For all of these syndromes it is important to follow the patients closely for the development of MDS/AML. And while HSCT can be curative in terms of the bone marrow defect, it is important to screen all potential family donors for the presence of the mutations in question. As Next-Gen sequencing of exomes and whole genomes are applied to more cases of unknown cytopenias with a propensity to develop MDS/AML, it is quite likely that more mutated genes will be identified to explain the phenotypes.
Acquired Aplastic Anemia and MDS/AML
Severe aplastic anemia (SAA) presents with pancytopenia and a hypocellular bone marrow. Although SAA may be the consequence of environmental and/or drug exposures as well as postinfectious, often postviral, hepatitis, many cases remain idiopathic. In such cases, a detailed diagnostic workup for germ line mutations and disorders associated with key BMF syndromes should be initiated. In addition, an important part of the differential diagnosis is hypoplastic MDS or hypoplastic refractory cytopenia of childhood, which may be difficult to distinguish from SAA. One study of 100 cases of SAA in children from the European Working Group of MDS in Childhood (EWOG-MDS) and the German SAA study group classified cases as MDS/RCC if patchy areas of erythropoiesis with abnormal maturation were observed in hypoplastic bone marrow.
138 Another report in children with SAA and MDS (RCC) noted that the histopathological pattern of MDS (RCC) was characteristically associated with islands of immature erythroid precursors and scattered granulocytic precursors along with the occasional micromegakaryocytes.
139 Examination of T-lymphocyte subsets has suggested that patients with low-risk MDS have increased T-helper and cytolytic T lymphocytes compared with patients with SAA.
140 Another potentially helpful discriminator is immunohistochemical staining for CD34, which has been considered to be higher in cases of hypoplastic MDS than in SAA.
141,142,143 However, this may not always be a distinguishing characteristic in RCC.
144 The existence of clonal, chromosomal abnormalities, such as monosomy 7, or specific gene mutations characteristic of MDS (see MDS section), can provide further evidence for a definitive diagnosis.
Approximately 10% to 15% of patients with idiopathic SAA who are not treated with allogeneic HSCT will develop MDS/AML. The frequency of developing MDS does not appear to vary when those patients with idiopathic versus viral-associated AA are compared.
145,146 In addition, a prospective study from Japan demonstrated that children with very severe presenting blood counts (neutrophil counts less than 200/µL) had a higher complete response rate than those with less severe presenting neutrophil counts, suggesting that those with more severe disease may have a more immunologically responsive disease and be less likely to have a primary MDS-like disorder.
147Children with SAA appear to develop MDS sooner than adults with SAA, and usually within the first 3 years following their diagnosis; this also suggests a fundamental difference in the disorders in children compared with adults. Another key point in managing such patients is that disease that is less responsive or requiring more prolonged periods of G-CSF treatment has a higher frequency of MDS/AML development.
148,149,150Allogeneic HSCT from a matched sibling donor (MSD) is a curative approach for children with newly diagnosed SAA and is also effective following the development of MDS.
151 Overall survival (OS) and event-free survival (EFS) for children with new-onset SAA transplanted with HLA-MSD have been reported to be approximately 90% and 86%, respectively.
152,153 Outcomes for children who develop MDS/AML after treatment for SAA have been reported to show 5-year OS of approximately 50% to 70%.
154,155Acquired amegakaryocytic thrombocytopenia (AAMT) can occur at any age and has been associated with environmental toxins, drugs, viral infections, and autoimmune disorders.
156,157,158 The disorder often will respond to a variety of immunosuppressive therapies.
159,160 Cases have been reported to progress to AML following development of a chromosome 5q2 abnormality.
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired hemolytic anemia due to increased sensitivity to complement-induced intravascular hemolysis. This increased sensitivity is secondary to a defect in glycophosphatidylinositol (GPI) anchoring of membrane proteins with decay-accelerating factor (DAF/CD55) and protectin (CD59/MIRL/MAC-IP) being affected. These cell-surface proteins play key roles in protecting red blood cells but also hematopoietic stem cells from destruction. The
PIGA gene, which encodes phosphatidylinositol glycan A, is the most commonly mutated enzyme. PNH may also evolve into SAA and then MDS/AML with the clonal evolution of additional chromosomal changes.
161,162
Environmental Mediated Mutagenesis
A large amount of evidence exists that certain environmental exposures can result in gene mutations and chromosomal rearrangements in bone marrow stem cells and can predispose individuals to the development of MDS and AML. Most of the evidence, however, has been derived from studies in adults. The very low incidence of MDS and AML in children has prevented large, definitive studies from being done. Nevertheless, there are several environmental exposures, both prenatal and postnatal, that have been examined and proven to be potentially important.
Ionizing radiation has been strongly associated with the development of MDS/AML. For example, an approximately 20-fold increase in AML was observed following the exposure of individuals to the atomic bombs on Nagasaki and Hiroshima during World War II. The peak of AML occurred between 6 to 8 years after exposure. The lack of a documented increase in leukemia in children prenatally exposed to the atomic bomb radiation is of note, and consistent with the lack of a definitive association of increased leukemia risk secondary to prenatal exposure to x-rays. In addition, there are no compelling data to link exposure to ultrasound or living near high-voltage power lines to the development of leukemia.
163,164,165,166However, maternal ingestion of various foods rich in topoisomerase II inhibitors, such as flavonoids in soy products and catechins in green and black tea, during pregnancy has been reported to be associated with an increased risk of AML with
MLL rearrangements in offspring.
167,168,169 Maternal alcohol consumption during pregnancy has also been linked to increased risk of AML in offspring in a dose-dependent fashion.
170 Maternal smoking during pregnancy has not been reported to increase the risk of AML in offspring,
170 although an association of cigarette smoking and AML has been linked in adults, thus emphasizing an additional rationale for smoking cessation.
171Exposure of individuals to environmental carcinogens, including pesticides, petroleum products, benzenes, and heavy metals, has been documented to increase the risk of MDS and AML. Of these, benzene has been the most definitively linked to the development of MDS/AML.
172,173 There is, however, evidence of an increased risk of leukemia in children in rural areas associated with increased exposure to pesticides
174 as well as an increased risk of AML in offspring of mothers exposed during gestation to significant levels of pesticides.
175 The observation of increased concentration of organophosphate pesticides in the children of farmworkers compared with adults suggests a possible reason for the increased leukemia risk in these children.
176 These associations should alert pediatric oncologists and pediatricians to include a careful history of potential environmental exposures in patients.
While environmental exposures to specific mutagens have been linked to the development of MDS/AML, the exposure of patients with cancer to ionizing radiation and chemotherapeutic agents also increases the risk of developing secondary leukemias. The risk and time to develop secondary MDS/AML is different depending on the type of exposure. For instance, secondary leukemias with characteristic
MLL gene rearrangements are primarily associated with exposure to drugs that function as topoisomerase inhibitors, including epipodophyllotoxins such as etoposide, and anthracyclines, such as doxorubicin, daunorubicin, idarubicin, and mitoxantrone.
177 This group of secondary AML usually develops within the first 3 to 5 years following exposure, although some cases have been reported 10 to 12 years later.
178 Both the cumulative dose and the schedule of such agents have been shown to be associated with the development of secondary AML
179; however, even single exposures have been reported in patients with subsequent secondary AML.
180,181Alkylating agents (e.g., cyclophosphamide, temozolomide) and related drugs (e.g., cisplatin) used primarily to treat patients with solid tumors represent a second class of chemotherapeutic agents that can result in secondary MDS/AML.
182,183 These secondary AML cases appear later, usually beyond 5 years of exposure, than those with
MLL rearrangements, and are more commonly associated with karyotypic abnormalities, such as monosomy 7.
Attempts to reduce the cardiotoxicity of anthracyclines have led to the use of cardioprotectant agents, such as dexrazoxane. The use of this cardioprotectant in combination with chemotherapy that included etoposide for patients with Hodgkin lymphoma led to the report of an increased incidence of secondary AML.
184 However, subsequent reports in patients with ALL and AML did not show an increased risk of secondary AML following the use of dexrazoxane.
185,186,187,188 Possible explanations for the differing results have included the schedule of dexrazoxane used in the Hodgkin lymphoma study, a distinct host effect in those with Hodgkin lymphoma, or chance variance based on small numbers.
189An additional, important component of increased risk to exogenous mutagens is the ability of the exposed person to metabolize or detoxify such compounds based on their genomic background. For example, polymorphisms in cytochrome p450 and glutathione-S-transferase, both of which are involved in the detoxification of environmental mutagens and chemotherapeutic agents, have been linked to increased risk of developing MDS/AML.
190,191,192 Specific polymorphisms of the thiopurine methyltransferase (
TPMT) gene that are key for the metabolism of 6-mercaptopurine and 6-thioguanine, both drugs used in the treatment of patients with ALL, have been reported to increase the risk of secondary MDS/AML.
193,194