Acute myeloid leukemia (AML) is a heterogeneous group of leukemias that result from clonal transformation of hematopoietic precursors through the acquisition of chromosomal rearrangements and multiple gene mutations. As a result of highly collaborative clinical research by pediatric cooperative cancer groups worldwide, disease-free survival has improved significantly during the past 3 decades. Further improvements in outcomes of children who have AML probably will reflect continued progress in understanding the biology of AML and the concomitant development of new molecularly targeted agents for use in combination with conventional chemotherapy drugs.
Acute myeloid leukemia (AML) is a heterogeneous group of leukemias that arise in precursors of myeloid, erythroid, megakaryocytic, and monocytic cell lineages. These leukemias result from clonal transformation of hematopoietic precursors through the acquisition of chromosomal rearrangements and multiple gene mutations. New molecular technologies have allowed a better understanding of these molecular events, improved classification of AML according to risk, and the development of molecularly targeted therapies. As a result of highly collaborative clinical research by pediatric cooperative cancer groups worldwide, disease-free survival (DFS) has improved significantly during the past 3 decades. Further improvements in the outcome of children who have AML probably will reflect continued progress in understanding the biology of AML and the concomitant development of new molecularly targeted agents for use in combination with conventional chemotherapy drugs.
Epidemiology and risk factors
Approximately 6500 children and adolescents in the United States develop acute leukemia each year. AML comprises only 15% to 20% of these cases but accounts for a disproportionate 30% of deaths from acute leukemia. The incidence of pediatric AML is estimated to be between five and seven cases per million people per year, with a peak incidence of 11 cases per million at 2 years of age. Incidence reaches a low point at age approximately 9 years, then increases to nine cases per million during adolescence and remains relatively stable until age 55 years. There is no difference in incidence between male and female or black and white populations. There is, however, evidence suggesting that incidence is highest in Hispanic children, intermediate in black children (5.8 cases per million), and slightly lower in white children (4.8 cases per million). The French-American-British (FAB) classification subtypes of AML are equally represented across ethnic and racial groups with the exception of acute promyelocytic leukemia (APL), which has a higher incidence among children of Latin and Hispanic ancestry.
During the years between 1977 and 1995, the overall incidence of AML remained stable, but there was a disturbing increase in the incidence of secondary AML as the result of prior exposure to chemotherapy and radiation. This risk remains particularly high among individuals exposed to alkylating agents (cyclophosphamide, nitrogen mustard, ifosfamide, melphalan, and chlorambucil) and intercalating topoisomerase II inhibitors, including the epipodophyllotoxins (etoposide).
Most children who have de novo AML have no identifiable predisposing environmental exposure or inherited condition, although a number of environmental exposures, inherited conditions, and acquired disorders are associated with the development of AML. Myelodysplastic syndrome and AML reportedly are associated with exposure to chemotherapy and ionizing radiation and also to chemicals that include petroleum products and organic solvents (benzene), herbicides, and pesticides (organophosphates).
A large number of inherited conditions predispose children to the development of AML. Among these are Down syndrome, Fanconi anemia, severe congenital neutropenia (Kostmann syndrome), Shwachman-Diamond syndrome, Diamond-Blackfan syndrome, neurofibromatosis type 1, Noonan syndrome, dyskeratosis congenita, familial platelet disorder with a predisposition to AML (FDP/AML), congenital amegakaryocytic thrombocytopenia, ataxia-telangiectasia, Klinefelter’s syndrome, Li-Fraumeni syndrome, and Bloom syndrome.
Finally, AML has been associated with several acquired conditions including aplastic anemia, myelodysplastic syndrome, acquired amegakaryocytic thrombocytopenia, and paroxysmal nocturnal hemoglobinuria.
Pathogenesis
AML is the result of distinct but cooperating genetic mutations that confer a proliferative and survival advantage and that impair differentiation and apoptosis. This multistep mechanism for the pathogenesis of AML is supported by murine models, the analysis of leukemia in twins, and the analysis of patients who have FDP/AML syndrome. Mutations in a number of genes that confer a proliferative and/or survival advantage to cells but do not affect differentiation (Class I mutations) have been identified in AML, including mutations of FLT3 , ALM , oncogenic Ras and PTPN11 , and the BCR/ABL and TEL/PDGFβR gene fusions. Similarly, gene mutations and translocation-associated fusions that impair differentiation and apoptosis (Class II mutations) in AML include the AML/ETO and PML/RARα fusions, MLL rearrangements, and mutations in CEBPA , CBF , HOX family members, CBP/P300 , and co-activators of TIF1 . AML results when hematopoietic precursor cells acquire both Class I and Class II genetic abnormalities. Although only one cytogenetic or molecular abnormality has been reported in many cases of AML, new molecular tools now are identifying multiple genetic mutations in such cases.
Accumulating data suggest that the leukemic stem cell arises at different stages of differentiation and involves heterogeneous, complex patterns of abnormality in myeloid precursor cells. The leukemic stem cell, also called the “self-renewing leukemia-initiating cell,” is located within both the CD34 + and CD34 − cell compartments and is rare (0.2–200 per 10 6 mononuclear cells). A recent study of pediatric AML suggested that patients who have FLT3 abnormalities in less mature CD34 + CD38 − precursor cells are less likely to survive than patients who have FLT3 mutations in more mature CD34 + CD38 + cells (11% versus 100% at 4 years; P = .002). Although sample sizes in this study were small, this result demonstrates the heterogeneity of genetic abnormalities in various stem cell compartments and suggests a worse outcome when less mature precursor cells harbor these abnormalities.
Pathogenesis
AML is the result of distinct but cooperating genetic mutations that confer a proliferative and survival advantage and that impair differentiation and apoptosis. This multistep mechanism for the pathogenesis of AML is supported by murine models, the analysis of leukemia in twins, and the analysis of patients who have FDP/AML syndrome. Mutations in a number of genes that confer a proliferative and/or survival advantage to cells but do not affect differentiation (Class I mutations) have been identified in AML, including mutations of FLT3 , ALM , oncogenic Ras and PTPN11 , and the BCR/ABL and TEL/PDGFβR gene fusions. Similarly, gene mutations and translocation-associated fusions that impair differentiation and apoptosis (Class II mutations) in AML include the AML/ETO and PML/RARα fusions, MLL rearrangements, and mutations in CEBPA , CBF , HOX family members, CBP/P300 , and co-activators of TIF1 . AML results when hematopoietic precursor cells acquire both Class I and Class II genetic abnormalities. Although only one cytogenetic or molecular abnormality has been reported in many cases of AML, new molecular tools now are identifying multiple genetic mutations in such cases.
Accumulating data suggest that the leukemic stem cell arises at different stages of differentiation and involves heterogeneous, complex patterns of abnormality in myeloid precursor cells. The leukemic stem cell, also called the “self-renewing leukemia-initiating cell,” is located within both the CD34 + and CD34 − cell compartments and is rare (0.2–200 per 10 6 mononuclear cells). A recent study of pediatric AML suggested that patients who have FLT3 abnormalities in less mature CD34 + CD38 − precursor cells are less likely to survive than patients who have FLT3 mutations in more mature CD34 + CD38 + cells (11% versus 100% at 4 years; P = .002). Although sample sizes in this study were small, this result demonstrates the heterogeneity of genetic abnormalities in various stem cell compartments and suggests a worse outcome when less mature precursor cells harbor these abnormalities.
Clinical presentation and diagnosis
The presentation of childhood AML reflects signs and symptoms that result from leukemic infiltration of the bone marrow and extramedullary sites. Replacement of normal bone marrow hematopoietic cells results in neutropenia, anemia, and thrombocytopenia. Children commonly present with signs and symptoms of pancytopenia, including fever, fatigue, pallor, bleeding, bone pain, and infections. Disseminated intravascular coagulation may be observed at presentation of all AML subtypes but is much more frequent in childhood APL. Infiltration of extramedullary sites can result in lymphadenopathy, hepatosplenomegaly, chloromatous tumors (myeloblastomas and granulocytic sarcomas), disease in the skin (leukemia cutis), orbit, and epidural space, and, rarely, testicular involvement. The central nervous system is involved at diagnosis in approximately 15% of cases. Patients who have high white blood cells counts may present with signs or symptoms of leukostasis, most often affecting the lung and brain.
A diagnosis is suggested by a complete blood cell count showing pancytopenia and blast cells and is confirmed by examination of the bone marrow. The diagnosis and subtype classification of AML is based on morphologic, cytochemical, cytogenetic, and fluorescent in situ hybridization analyses, flow cytometric immunophenotyping, and molecular testing (eg, FLT3 mutation analysis).
Treatment of childhood acute myeloid leukemia
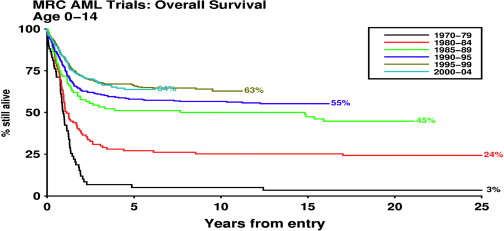
The prognosis of children who have AML has improved greatly during the past 3 decades ( Fig. 1 ). Rates of complete remission (CR) as high as 80% to 90% and overall survival (OS) rates of 60% now are reported. ( Table 1 ) This success reflects the use of increasingly intensive induction chemotherapy followed by postremission treatment with additional anthracyclines and high-dose cytarabine or myeloablative regimens followed by stem cell transplantation (SCT). The drugs used in the treatment of AML have changed little, but refinement of their delivery and striking advances in supportive care have allowed administration of optimally intensive therapy with less morbidity and mortality. Better postrelapse salvage therapy also has contributed to the improvement in OS.
| Study (Years of Enrollment) | Number of Patients Enrolled | Non-Responders (%) | Early Death Rate (%) | Complete Response (%) | % 5-Year Event-free Survival (SE) | % 5-Year Overall Survival (SE) | Death Rate in Complete Response (%) | Cumulative Doses of ara-C, Etoposide, and Anthracyclines a | % of Total Number of Patients Who Underwent Allogeneic Stem cell Transplantation |
|---|---|---|---|---|---|---|---|---|---|
| AIEOP92 (1992–2001) | 160 | 5 | 6 | 89 | 54 (4) | 60 (4) | 7 | No strict protocol guidelines | 29 |
| AML-BFM93 (1993–1998) | 427 | 10 | 7 | 83 | 51 (3) | 58 (2) | 4 | 41.1 g/m 2 | 7 |
| 950 mg/m 2 | |||||||||
| 300–400 mg/m 2 | |||||||||
| CCG2891 (1989–1995) | 750 | 18 | 4 | 78 | 34 (3) | 47 (4) | 15 | 14.6 g/m 2 | 25 |
| 1100 mg/m 2 | |||||||||
| 180 mg/m 2 | |||||||||
| DCOG-ANLL 92/94 (1992–1998) | 78 | 8 | 10 | 82 | 42 (6) | 42 (6) | 16 | 33.2 g/m 2 | 27 |
| 950 mg/m 2 | |||||||||
| 400 mg/m 2 | |||||||||
| EORTC-CLG 58,921 (1993–2000) | 166 | 13 | 2 | 84 | 48 (4) | 62 (4) | 6 | 23.32–29.32 g/m 2 | 20 |
| 1350 mg/m 2 | |||||||||
| 380 mg/m 2 | |||||||||
| GATLA-AML90 (1990–1997) | 179 | 11 | 20 | 70 | 31 (4) | 41 (4) | 7 | 41.1 g/m 2 | 3 |
| 1450 mg/m 2 | |||||||||
| 300 mg/m 2 | |||||||||
| LAME91 (1991–1998) | 247 | 5 | 4 | 91 | 48 (4) | 62 (4) | 6 | 9.8–13.4 g/m 2 | 30 |
| 400 mg/m 2 | |||||||||
| 460 mg/m 2 | |||||||||
| NOPHO-AML93 (1993–2001) | 223 | 5 | 2 | 92 | 50 (3) | 66 (3) | 2 | 49.6–61.3 g/m 2 | 25 |
| 1600 mg/m 2 | |||||||||
| 300–375 mg/m 2 | |||||||||
| PINDA-92 (1992–1998) | 151 | 5 | 26 | 68 | 36 | 36 | 4 | 7.64 g/m 2 | — |
| 450 mg/m 2 | |||||||||
| 350 mg/m 2 | |||||||||
| POG8821 (1988–1993) | 511 | 19 | 4 | 77 | 31 (2) | 42 (2) | 8 | 55.7 g/m 2 | 13 |
| 2250 mg/m 2 | |||||||||
| 360 mg/m 2 | |||||||||
| PPLLSG98 (1998–2002) | 104 | 13 | 8 | 80 | 47 (5) | 50 (5) | 10 | 7.0–15.1 g/m 2 | Not reported |
| 450–950 mg/m 2 | |||||||||
| 420–600 mg/m 2 | |||||||||
| St Jude-AML91 (1991–1997) | 62 | 16 | 3 | 79 | 44 (15) | 57 (11) | ? | 3.8 g/m 2 | Not given |
| 1200 g/m 2 | |||||||||
| 270 mg/m 2 | |||||||||
| UK MRC AML10 (1988–1995) | 303 | 3 | 4 | 93 | 49 | 58 | 10 | 10.6 g/m 2 | 20 |
| 500–1500 mg/m 2 | |||||||||
| 550 mg/m 2 | |||||||||
| UK MRC AML12 (1995–2002) | 455 | 4 | 4 | 92 | 56 | 66 | 6 | 4.6–34.6 g/m 2 | 8 |
| 1500 mg/m 2 | |||||||||
| 300–610 mg/m 2 |
a Cumulative dose of anthracyclines was calculated by applying the following arbitrary conversion factors to obtain daunorubicin equivalents: idarubicin, 5×; mitoxantrone, 5×; doxorubicin, 1×. Some groups (Leucemie Aique Myeloide Enfant and the Medical Research Council in the United Kingdom) also administered amsacrine, which is not included in calculated total anthracycline exposure.
Treatment of AML in children generally is based on an anthracycline, cytarabine, and etoposide regimen given as a minimum of four cycles of chemotherapy. A recent report compared the results of anthracycline, cytarabine, and etoposide regimens used by 13 national study groups. The regimens differed in many ways, including the cumulative doses of drugs, the choice of anthracycline, the number and intensity of blocks of treatment, and the intrathecal chemotherapy used for central nervous system (CNS) prophylaxis. Treatment generally was risk stratified, although the definition of risk groups varied, as did the indications for SCT. Despite the varying strategies, results are relatively similar (see Table 1 ). Many groups now achieve CR rates of 80% to 90%, relapse rates of 30% to 40%, event-free survival (EFS) rates of 50%, and OS rates of 60%.
Because of the small number of pediatric patients who have AML, many important questions have not been addressed in the context of randomized trials. The unresolved issues include the optimal intensity of chemotherapy, the optimal anthracycline, the optimal dose of cytarabine, the cumulative dose of anthracycline that minimizes cardiotoxicity without compromising outcome, the role of allogeneic SCT in first CR, and the use of risk-directed therapy.
Induction and Consolidation Therapy
The most favorable outcomes are achieved by the use of a relatively high cumulative dose of either anthracycline or cytarabine (see Table 1 ). The schedule and timing of intensification also are important. The Children’s Cancer Group (CCG) reported that intensively timed induction therapy (the second cycle delivered 10 days after the first cycle) was more advantageous than standard therapy (the second cycle delivered 14 or more days after the first cycle, dependent on bone marrow status and cell-count recovery). Both the CR and EFS rates were significantly higher with intensively timed dosing, regardless of postremission therapy, suggesting that the depth of remission may profoundly affect survival. The benefit derived from early intensification, whether achieved by time sequencing or by adjusting cytarabine and etoposide doses to achieve a targeted plasma level, may be lost, however, if prolonged neutropenia and thrombocytopenia cause unacceptable delays in subsequent treatment. The intensification of early therapy beyond a certain threshold therefore is unlikely to improve outcome and may even be detrimental to OS.
In a Medical Research Council (MRC) study, an additional course of postremission chemotherapy (four versus five courses in total) provided no advantage to patients already receiving intensive treatment, suggesting a plateau in the benefit of conventional postremission chemotherapy. If such a plateau is confirmed, it is likely that any additional antileukemic effect will have to come from alternative approaches, such as targeted or cellular therapies.
Certain anthracyclines are favored for their perceived greater antileukemic effect and/or their lower cardiotoxicity, but no anthracycline agent has been demonstrated to be superior. The MRC found daunorubicin and mitoxantrone to be equally efficacious but mitoxantrone to be more myelosuppressive. Idarubicin is used commonly because in vitro and preclinical studies suggest that it offers a greater clinical benefit because of its faster cellular uptake, increased retention, and lower susceptibility to multidrug resistant glycoprotein. In addition, its main metabolite, idarubicinol, has a prolonged plasma half-life (54 hours) and has antileukemic activity in the cerebrospinal fluid. In the Berlin-Frankfurt-Münster (BFM) AML 93 trial, induction therapy with idarubicin, cytarabine, and etoposide (AIE) resulted in significantly greater blast-cell clearance at day 15 than induction with daunorubicin, cytarabine, and etoposide (ADE) ( P = .01) but did not improve 5-year OS (51% with AIE versus 50% with ADE; P = .72) or EFS (60% for AIE versus 57% for ADE; P = .55). Similarly, the Australian and New Zealand Children’s Cancer Study Group reported that idarubicin and daunorubicin were equally efficacious, but idarubicin was more toxic. The addition of cyclosporin A to induction chemotherapy to inhibit P-glycoprotein–mediated anthracycline efflux did not prolong the duration of remission or improve OS in children.
Another important question is whether the cumulative dose of anthracyclines can be reduced safely without compromising survival. Although cumulative doses above 375 mg/m 2 increase the risk of cardiotoxicity, EFS is lower in protocols that use lower doses of anthracycline. Optimal results may be achievable with a cumulative dose of approximately 375 to 550 mg/m 2 if high-dose cytarabine is used in postremission therapy. The full impact of cardiotoxicity, particularly late cardiotoxicity, also is poorly defined. In the MRC AML10 protocol, which delivered a high cumulative anthracycline dose (550 mg/m 2 ), 9 of 341 registered patients died of acute cardiotoxicity (all after a cumulative dose of 300 mg/m 2 ); 7 of the 9 deaths occurred during an episode of sepsis. Subclinical deficits in cardiac function would have gone undetected in the absence of cardiac monitoring. Minimizing cardiotoxicity is important, however, and cardioprotectant agents and liposomal anthracyclines with reduced cardiotoxicity are being tested.
The use of high-dose cytarabine in postremission therapy seems to be important in improving survival, but the optimal dose has not been determined. Core binding factor (CBF) leukemias may respond particularly well to multiple courses of high-dose cytarabine.
Central Nervous System–directed Therapy
The impact of CNS involvement on EFS is not well defined. Most pediatric clinical trial groups use intrathecal chemotherapy for CNS prophylaxis, employing either one or three agents and various doses. Not all pediatric groups routinely use intrathecal CNS prophylaxis, however, and few adult groups do. The correlation between the type of CNS treatment given and the incidence of CNS relapse is not clear. The CNS relapse rate seems to be around 2% for isolated CNS relapse and between 2% and 9% for combined CNS and bone marrow relapse. The low rate of CNS relapse may reflect both the use of intrathecal chemotherapy and the CNS protection afforded by high-dose cytarabine and by idarubicin, both of which can penetrate the CNS. Cranial irradiation, because of its sequelae, is not widely used as prophylaxis. It is used currently only by the BFM Study Group, which observed an increase in CNS and systemic relapse in patients who did not receive cranial irradiation in the AML BFM 87 trial. The current AML BFM 98 trial is exploring reduction of the dose of cranial irradiation to limit late sequelae. The necessity of cranial irradiation for patients who have CNS involvement at presentation or CNS relapse is unproven. Many groups reserve cranial irradiation for patients whose CNS is not cleared of leukemic cells by intrathecal and intensive systemic chemotherapy.
Maintenance Therapy
Maintenance therapy is no longer used in the treatment of AML, having failed to demonstrate benefit except in BFM studies. Patients who have APL, however, do seem to benefit from antimetabolite maintenance treatment given with all-trans retinoic acid (ATRA). In patients who have non-APL AML, maintenance treatment showed no benefit in two randomized studies (Leucemie Aigue Myeloblastique Enfant 91 and CCG 213); these studies even suggested that maintenance therapy may be deleterious when intensive chemotherapy is used and may contribute to clinical drug resistance and treatment failure after relapse.
Stem Cell Transplantation
SCT is the most successful curative treatment for AML; it produces a strong graft-versus-leukemia effect and can cure even relapsed AML. Its potential benefit, however, must be weighed against the risk of transplantation-related mortality and the late sequelae of transplantation. SCT has become a less attractive option as the outcomes of increasingly intensive chemotherapy and postrelapse salvage therapy have improved. Furthermore, although SCT is reported to provide a survival advantage for patients in first CR, studies so far have used matched sibling donors, who are available to only about one in four patients. Although experienced groups have reported comparable outcomes with alternative donors, it is too early to determine whether their wider use will result in greater transplantation-related mortality.
The role of allogeneic SCT, particularly whether it should be done during first CR or reserved for second remission, remains the most controversial issue in pediatric AML. Competing factors, particularly risk group, may tip the balance in favor of SCT or intensive chemotherapy. Most groups agree that children who have APL, AML and Down syndrome or AML and the t(8;21) or inv(16) are not candidates for SCT in first CR, but opinions differ about patients in the standard-risk and high-risk categories. The trend in Europe is to reduce the use of SCT in first CR, but in the United States SCT in first CR is supported. Both views have been reported recently.
In the absence of randomized, controlled trials comparing allogeneic SCT with postremission intensive chemotherapy, “biologic randomization” or “donor versus no donor” studies are accepted as the least biased comparison methods, but even these are open to criticism. Much of the trial data used to support the benefits of SCT and intensive chemotherapy are old and do not reflect current improvements in SCT and intensive chemotherapy. A meta-analysis of studies enrolling patients younger than 21 years of age between 1985 and 2000 that recommended SCT if a histocompatible family donor were available found that SCT from a matched sibling donor reduced the risk of relapse significantly and improved DFS and OS.
The MRC AML10 (included in the meta-analysis) and AML12 studies combined (relapse risk did not differ between the trials; P = .3) showed a significant reduction in relapse risk ( 2P = 0.02) but no significant improvement in DFS ( 2P = 0.06) or OS ( 2P = 0.1). MRC AML10 is typical of a number of trials in which SCT significantly reduced the risk of relapse, but the resulting improvement in survival was not statistically significant (68% versus 59%; P = .3). The small number of pediatric patients in AML10 hinders meaningful interpretation, but at 7 years’ follow-up SCT recipients (children and adults) who had a suitable donor showed a significant reduction in relapse risk (36%, versus 52% in patients who did not have a suitable donor; P = .0001) and a significant improvement in DFS (50%, versus 42% in patients who did not have a suitable donor; P = .001) but no significant improvement in OS (55% versus 50%; P = .1). The reduction in relapse risk was seen in all risk and age groups, but the significant benefit in DFS was seen only in the cytogenetic intermediate-risk group (50% versus 39%; P = .004). The 86 children who had a donor, 61 of whom (71%) underwent SCT, had no survival advantage, and children who did not undergo SCT were salvaged more easily.
The lack of benefit found for pediatric SCT in the MRC trials mirrors the experience of the BFM. CCG trial 2891, however, showed a significant survival advantage for patients who underwent allogeneic SCT versus autologous SCT (60% versus 53%; P = .002) or chemotherapy (60% versus 48%; P = .05) as postremission treatment, although autologous SCT provided no advantage over intensive chemotherapy. The benefit was most marked in patients who had received intensively timed induction chemotherapy. The CCG analysis was not a true intent-to-treat comparison, however. Although it included patients whether or not they received SCT, it did not include all patients who lacked a donor; instead, it included only patients who lacked a donor and who were randomly assigned to autologous SCT instead of chemotherapy, and favorable cytogenetics were overrepresented among patients who had a donor (38% versus 23%). The MRC AML10 (5-year OS, 58%) and CCG 2891 (5-year OS, 47%; 49% for the intensive arm) studies enrolled patients during approximately the same time period, although the patient populations may not have been comparable. It is possible that the improved outcomes achieved by intensive chemotherapy may diminish the role of SCT in first CR of AML and that SCT provides a benefit only when compared with relatively less intensive treatment.
Randomized studies analyzed according to intent to treat have failed to show that autologous SCT provides a survival advantage over intensive chemotherapy, and a meta-analysis concluded that data were insufficient to determine whether autologous SCT is superior to nonmyeloablative chemotherapy.
The controversy continues. In some groups, all patients who have a matched sibling donor proceed to SCT, whereas in others SCT is reserved for patients at high risk, although high risk is not defined consistently. In the MRC, SCT has not been demonstrated to reduce the risk of relapse even in children at high risk. Unless it is demonstrated to reduce the risk of relapse, transplantation can offer no benefit. SCT may have a role in the treatment of pediatric AML in first CR if the graft-versus-leukemia effect can be expanded by pre- and posttransplantation graft manipulation, which may include the use of killer-cell immunoglobulin receptor–incompatible donors and donor lymphocyte infusions.
There is also a need to improve risk-group stratification and to identify better the children who may benefit from SCT. This goal may be achieved by identifying better prognostic indicators and by using minimal residual disease (MRD) monitoring, both of which are discussed in later sections.
Special Subgroups
Acute myeloid leukemia in children who have Down syndrome
Children who have Down syndrome who develop AML generally do so between 1 and 4 years of age. This subset of cases of AML is very responsive to therapy but carries a significant risk of early mortality. Children treated during the past decade have had a reported EFS estimate of 83%, with relapse rates as low as 3%, The recommendation is to limit the cumulative anthracycline dose to 240 to 250 mg/m 2 or to reduce overall dose intensity rather than the absolute dose.
Acute promyelocytic leukemia
Children who have APL are treated with special APL protocols that combine ATRA with intensive chemotherapy. Although ATRA can cause considerable (but manageable) toxicity in some children, this approach induces a stable and continuous remission without the early hemorrhagic deaths that previously characterized this type of leukemia. APL is the only subtype of AML in which maintenance chemotherapy is believed to be of benefit. SCT in first CR is not indicated for a disease that responds so well to chemotherapy. Regimens increasingly based on alternatives to traditional chemotherapy, including ATRA and arsenic trioxide, are being tested.
Relapsed acute myeloid leukemia
After relapse, chemotherapy alone is unlikely to be curative, and the survival rate is only 21% to 33% in recent reports. In these reports, the length of first remission was the best predictor of survival. Various remission induction regimens, including fludarabine plus cytarabine and mitoxantrone plus cytarabine, seem to give similar results. The addition of liposomal daunorubicin to fludarabine plus cytarabine is being tested currently to try to improve CR rates while minimizing cardiotoxicity. It is important to reduce the toxicity of reinduction to a level that allows SCT to proceed, because children who receive SCT can have a 5-year survival probability of 60% (56% after early relapse; 65% after late relapse).
The targeted immunotherapy agents gemtuzumab ozogamicin and clofarabine have shown activity against relapsed AML. Gemtuzumab ozogamicin has been shown to be safe and well tolerated in children and, as a single agent, has induced responses in 30% of patients who have recurrent CD33 + AML. Clofarabine has demonstrated activity against refractory and relapsed AML. Both of these drugs may be more useful when given in combination with other chemotherapeutic agents.
A second allograft seems to offer a benefit to patients who experience relapse after SCT during first CR. Despite a high rate of transplantation-related mortality and second relapse, more than one third of patients are reported to be long-term survivors. Patients who undergo SCT during remission may have an even better outcome. Therefore every effort should be made to induce remission before the second SCT.
Prognostic Factors
Although clinical measures of tumor burden, such as leukocyte count and hepatosplenomegaly, largely have been replaced by genetic factors in the risk-classification schemes of contemporary treatment protocols, several clinical features are still prognostically important. In both adult and pediatric patients who have AML, age at diagnosis is associated inversely with the probability of survival. In an analysis of 424 patients less than 21 years of age, an age greater than 10 years at diagnosis was significantly associated with a worse outcome, even after controlling for cytogenetics, leukocyte count, and FAB subtype. The effect of age was important only among patients treated in contemporary trials, reinforcing the view that the effect of any prognostic factor ultimately depends on the therapy given. Two recent studies suggest that another clinically apparent feature—ethnicity—may be an important predictor of outcome. Among more than 1600 children who had AML treated on the CCG 2891 and 2961 trials, black children treated with chemotherapy had a significantly worse outcome than white children treated with chemotherapy, a disparity that the authors suggest may reflect pharmacogenetic differences. Body mass index, another easily measured clinical feature, also may affect the outcome of children who have AML. In the CCG 2961 trial, underweight and overweight patients were less likely to survive than normoweight patients because of a greater risk of treatment-related death.
In addition to clinical features, certain pathologic features, such as M0 and M7 subtypes, seem to carry prognostic importance in AML. The present authors and others have demonstrated that non–Down syndrome patients who have megakaryoblastic leukemia have significantly worse outcomes than patients who have other subtypes of AML. The EFS estimates for patients who have megakaryoblastic leukemia treated in the CCG 2891 trial or in the St Jude trial were only 22% and 14%, respectively. In the St Jude study and in a report from the European Group for Blood and Marrow Transplantation, patients who underwent SCT during first remission had a better outcome than those who received chemotherapy, suggesting that SCT should be recommended for these patients. A study by French investigators, however, suggested that children who had megakaryoblastic leukemia with the t(1;22), but without Down syndrome, had a better outcome than similar children who did not have the t(1;22), indicating that this subgroup may not need transplantation. In addition, the BFM study group reported an improved outcome for patients who had megakaryoblastic leukemia treated in recent, more intensive trials. SCT did not provide a benefit to patients treated in these trials. Thus, the role of SCT for patients who have megakaryoblastic leukemia remains controversial.
Conventional cytogenetic studies have demonstrated that the karyotype of leukemic blast cells is one of the best predictors of outcome. An analysis of more than 1600 patients enrolled in the MRC AML 10 trial revealed that t(8;21) and inv(16) were associated with a favorable outcome (5-year OS estimates, 69% and 61%, respectively), whereas a complex karyotype, -5, del(5q), -7, and abnormalities of 3q predicted a poor outcome. On the basis of these observations, the MRC investigators proposed a cytogenetics-based risk classification system that is used by many cooperative groups today. Among the 340 patients in the MRC study who were less than 15 years old, those with a favorable karyotype had a 3-year survival estimate of 78%, compared with 55% for the intermediate-risk group and 42% for the high-risk group. Other cooperative groups have confirmed the MRC findings, with slightly different results for some subgroups that probably reflect differences in therapy. For example, in the Pediatric Oncology Group 8821 trial, patients who had t(8;21) had a 4-year OS estimate of 52% and those who had inv(16) had an estimate of 75%. Similarly, among adults who had AML treated in Cancer and Leukemia Group B trials, patients who had these karyotypes had a better outcome than others and had a particularly good outcome when treated with multiple courses of high-dose cytarabine.
Because both t(8;21) and inv(16) disrupt the CBF, they are often referred to as “CBF leukemias” and are grouped together in risk-classification systems. Several studies, however, have demonstrated that CBF leukemia is a heterogeneous group of diseases in adults and therefore probably is heterogeneous in children as well. An analysis of 312 adults who had CBF AML demonstrated that, although CR and relapse rates were similar for patients who had t(8;21) and inv(16), OS was significantly worse for those who had t(8;21), primarily because of a lower salvage rate after relapse. In addition, race was prognostically important among patients who had t(8;21), whereas sex and secondary cytogenetic changes were predictive of outcome among patients who had inv(16). A similar analysis of 370 adults who had CBF AML confirmed the heterogeneity of this type of AML and confirmed the poor outcome after relapse among patients who had t(8;21). Not surprisingly, in both studies, outcome depended on treatment intensity.
Other prognostically important cytogenetic abnormalities include rearrangements of the MLL gene, located at chromosome band 11q23. The abnormality is usually a reciprocal translocation between MLL and one of more than 30 other genes in distinct chromosomal loci. MLL rearrangements are seen in as many as 20% of cases of AML, although the reported frequency varies among studies. In general, children and adults whose leukemic cells contain 11q23 abnormalities are considered at intermediate risk, and their outcome does not differ significantly from that of patients without these translocations (3-year OS estimate, 50% in the MRC AML 10 trial). Some studies, however, suggest that t(9;11) confers a favorable outcome. Among patients treated for AML at St Jude, those who had t(9;11) had a better outcome (5-year EFS estimate, 65%) than did patients in all other cytogenetic or molecular subgroups. This finding may be attributable to the use of epipodophyllotoxins and cladribine, both of which are effective against monoblastic leukemia.
In the MRC AML 10 study mentioned previously, monosomy 7 was associated with a particularly poor outcome (5-year OS, 10%) but was detected in only 4% of cases.
Because of the rarity of this abnormality, an international collaborative study was undertaken to characterize further the impact of -7 and del(7q) in children and adolescents who have AML. In this study, which included 172 patients who had -7 (with or without other abnormalities) and 86 patients who had del(7q) (also with or without other changes), patients who had -7 had lower CR rates (61% versus 89%) and worse outcome (5-year survival, 30% versus 51%) than those who had del(7q). Patients who had del(7q) and a favorable genetic abnormality had a good outcome (5-year survival, 75%), suggesting that the del(7q) does not alter the impact of the favorable feature. By contrast, patients who had -7 and inv(3), -5/del(5q), or +21 had a dismal outcome (5-year survival, 5%) that was not improved by SCT.
During the past 10 years, molecular studies have demonstrated heterogeneity within cytogenetically defined subgroups of AML and have identified new, prognostically important subgroups. Mutations of c-kit , ras , and FLT3 have been detected in cases of childhood and adult AML; c-kit mutations may be particularly important in cases of CBF leukemia. Several studies demonstrated that among adult patients who had t(8;21), those who had mutations at c-kit codon 816 had a significantly higher relapse rate and worse outcome than those who had wild-type c-kit . In some studies, mutations of c-kit also seem to confer a worse outcome among patients who have inv(16). Although c-kit mutations have been detected in 3% to 11% of pediatric AML cases, their prognostic impact is uncertain. One study found c-kit mutations in 37% of cases of CBF leukemia, but these cases did not differ from others in outcome. In contrast, the Japanese Childhood AML Cooperative Study Group found that c-kit mutations, in 8 of 46 patients who had t(8;21), were associated with significantly worse OS, DFS, and relapse rates.
The impact of FLT3 mutations in childhood and adult AML has been established by dozens of studies, only a few of which are summarized here. In one of the first studies reported, the estimated 5-year OS rate was only 14% for adult patients who had internal tandem duplications (ITD) of FLT3 , and the presence of these mutations was the strongest prognostic factor in multivariate analysis. Similarly, in an analysis of 106 adults who had AML treated in MRC trials, 13 of the 14 patients who had FLT3 ITD died within 18 months of diagnosis. A subsequent study of 854 patients treated in the MRC AML trials demonstrated a FLT3 ITD, present in 27% of cases, was associated with an increased risk of relapse and a lower probability of DFS, EFS, and OS. Other reports have confirmed the presence of FLT ITD in 20% to 30% of adult AML cases, but some studies suggest that its negative prognostic impact may depend on the absence of the wild-type allele or the ratio of the mutant to the wild-type allele.
Studies of childhood AML identify FLT3 ITD in only 10% to 15% of cases, but still it is associated with a poor outcome. Among 91 pediatric patients who had AML treated in CCG trials, the 8-year EFS estimate was only 7% for patients who had FLT3 ITD, whereas among 234 patients treated on Dutch AML protocols, the 5-year EFS for these patients estimate was only 29%. In both studies, multivariate analysis demonstrated that FLT3 -ITD was the strongest predictor of relapse. A more recent study of 630 patients treated in contemporary CCG trials confirmed the poor outcome associated with FLT3 ITD and demonstrated that survival decreased with an increasing allelic ratio of FLT ITD to FLT3 wild-type. The estimated progression-free survival was considerably lower with a ratio greater than 0.4 than with a lower ratio (16% versus 72%). CCG investigators also compared the outcome of patients who had FLT3 ITD in CD34 + /CD33 − precursors with that of patients who had the mutated gene in only the more mature CD34 + /CD33 + progenitors. Patients who had the mutation in the less mature precursors had dramatically worse outcomes, confirming the heterogeneity within FLT3 ITD–positive cases of AML and suggesting that only a subset of these patients have a poor prognosis. Data from studies by the Pediatric Oncology Group suggest that gene expression profiles also may be used to identify patients who have a good prognosis despite FLT3 mutations.
Other molecular alterations reported to be prognostic factors in AML include expression of ATP-binding cassette transporters, CEBPA mutations, DCC expression, secretion of vascular endothelial growth factor, expression of apoptosis-related genes, expression of BAALC , expression of ERG , NPM1 mutations, partial tandem duplications (PTD) of the MLL gene, and global gene expression patterns. The clinical relevance of these alterations has been reviewed comprehensively and is discussed only briefly here. Mutations of the nucleophosmin member 1 ( NPM1 ) gene have been detected in about 50% of cases of adult AML with a normal karyotype but occur much less commonly in childhood AML. In both populations, NPM1 mutations are associated with FLT3 ITD; however, in patients who have wild-type FLT3 , NPM1 mutations are associated with a favorable outcome. MLL PTD occur in about 5% to 10% of adult AML cases and, like NPM1 mutations, commonly are associated with FLT3 ITD. MLL PTD seem to be an adverse prognostic factor, but it is not clear whether the negative impact is related to the association with FLT3 ITD. High expression of the BAALC gene and the ERG gene are additional factors that have independent negative prognostic significance among adult patients who have a normal karyotype, whereas mutations of the CEBPA gene are associated with a favorable outcome. A risk-classification scheme for adults who have a normal AML karyotype that incorporates the status of FLT3 , NPM1 , BAALC , MLL , and CEBPA has been proposed and may be used in future clinical trials. MLL PTD, BAALC , and CEBPA have not been studied extensively in childhood AML. Nevertheless, it is likely that forthcoming pediatric clinical trials will use gene-expression profiling to identify important prognostic subgroups that may benefit from more intensive or novel therapies.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


