Acute Lymphoblastic Leukemia and Lymphoma
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is a highly aggressive neoplasm of hematopoietic cells of lymphoid lineage. Collections of abnormal T- or B lymphoblasts may be found in the bone marrow, peripheral blood, and other extramedullary sites. ALL is predominantly a childhood cancer, with two-thirds of new cases diagnosed in children younger than 15 years of age. ALL was uniformly fatal until the 1960 but, due to advances in chemotherapy and supportive care, is now cured in over 80% of children. Adults diagnosed with ALL, in contrast, have a poor overall prognosis. Important factors in assessing prognosis are the age of the patient, type of lymphoid cell involved (T cell vs B cell), and the presence of high-risk cytogenetic markers, such as the t(9;22) (BCR–ABL) translocation. Burkitt’s lymphoma, a malignancy of mature B cells, has been historically classified as a B-ALL due to its high-grade leukemia-like features but is both diagnostically and prognostically a separate entity from precursor B-ALL. Burkitt’s lymphoma is addressed in the chapter on non-Hodgkin lymphomas.
• ALL is the most common malignancy of childhood.
• Childhood ALL has a much better prognosis than adult ALL.
• Highly aggressive lymphoid malignancy with B-cell (80%) and T-cell (20%) immunophenotypes.
• Certain cytogenetic alterations, such as t(9;22), are associated with inferior prognosis while others, such as t(12;21), are associated with favorable outcomes.
EPIDEMIOLOGY AND ETIOLOGY
Leukemia comprises 32% of malignancies in children younger than 15 years. Of these, the majority are ALL. Each year approximately 2400 children in the United States are diagnosed with ALL. The peak incidence in children is between ages 2 and 5. Leukemia rates are significantly higher in Caucasian children, with a nearly threefold higher incidence over African-American children. ALL is almost 30% more common in males than females. Overall, the incidence of childhood ALL has increased in the past 20 years at a rate of 0.9% per year. Adult ALL is less common, with approximately 1000 new cases diagnosed per year. The incidence of ALL decreases from age 15 until 50; then a second, minor increase in new cases appears. A third peak appears at age 80. The lifetime risk of developing ALL is 0.13%, or approximately 1 in 789 men and women (1).
Several reports have suggested that inadvertent exposure to radiation in utero and postnatal radiation treatment for such conditions as tinea capitis and thymic enlargement increase the risk of ALL (2). A common cytogenetic translocation involving ETV-6 was retrospectively detected in neonatal blood spots of children who were diagnosed with ALL between ages 2 and 5, suggesting that ALL can be initiated by somatic translocation in utero but requires additional molecular events to fully develop (3). Limited and/or inconsistent evidence links ALL to parental smoking, infection, diet, electromagnetic fields, hydrocarbons and, possibly, radiation delivered during the course of diagnostic studies such as CT scans (4).
The following characteristics are associated with ALL:
• Male sex
• Age 2–5
• Caucasian race
• Higher socioeconomic status (SES)
• Hereditary factors (Down syndrome, Bloom syndrome, ataxia telangectasia, neurofibromatosis, Klinefelter syndrome, Shwachman syndrome, and Langerhans cell histiocytosis)
• Radiation and chemical exposure is controversial but may increase overall risk of ALL both in utero and during childhood
• Overall likelihood of developing ALL in one’s lifetime is 0.13%, or 1 in 789
ALL CLASSIFICATION
Proper characterization of the specific hematopoietic lineage involved in ALL is crucial for assessing risk and for treatment. ALL may be classified according to the presence or absence of various cell surface and intracellular markers.
Both immunohistochemistry and flow cytometry may be used to identify expression of cell surface and cytoplasmic proteins. These techniques use panels of lineage-specific antibodies directed against B-lymphoid and T-lymphoid antigens to stain patient bone marrow and lymph node samples. Common immunophenotypes are presented in Table 28-1.
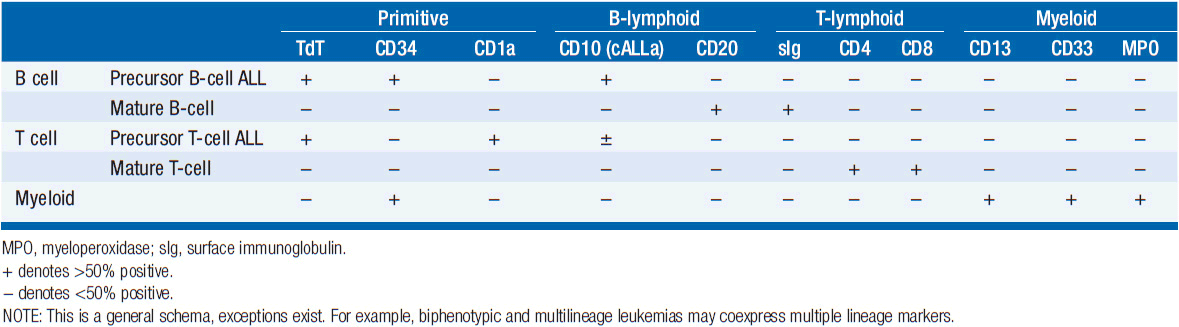
Both B and T lymphoblasts typically express terminal deoxytransferase (TdT) and/or the primitive hematopoietic cell surface marker CD34. Approximately 80% of adult ALL patients have B-ALL, while 5% have Burkitt’s (an aggressive tumor of peripheral follicular B cells) and the remainder are precursor T-cell ALL.
Precursor B-ALL cells express CD19 and at least one other B-lineage marker such as CD20, CD24, CD22, CD21, or CD79. More than 90% also express CD10, a marker known as CALLA, or common ALL antigen. In addition, 25% of patients have cytoplasmic Ig staining.
Precursor T-cell leukemias express CD7, TdT, and cytoplasmic CD3 antigen. Expression of CD1a is highly characteristic of T-ALL. More highly differentiated thymocytes acquire CD2 and CD5 and, later, CD4 and CD8. Mature thymocytes express functional T-cell receptor (TCR) and surface CD3. TCR rearrangement studies may be conducted to establish clonality.
As in adults, approximately 80% of children with ALL too have B-ALL, whereas 2% have mature B-cell (Burkitt’s) leukemia/lymphoma and 15% T-ALL.
• ALL may be classified as B- or T-cell origin using intracellular and cell surface markers.
• B-ALL markers most commonly include: CD10, CD19, CD20, CD21, CD22, CD24, and CD34 and TdT. Twenty-five percent of B-ALL will have cytoplasmic Ig staining.
• T-ALL markers include CD7, TdT, CD1a, and cytoplasmic CD3. More differentiated leukemia markers of T-cell origin include CD2, CD5, and CD4 or CD8.
DIAGNOSIS OF ALL
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
Children with ALL may have an insidious or explosive course before diagnosis, whereas adults present more uniformly with rapid-onset disease. Physical signs and symptoms are the result of marrow failure from leukemia cell proliferation.
Patients commonly present with signs and symptoms of anemia, such as pallor, fatigue, lethargy, and, in adults, cardiac angina. Thrombocytopenia, another common sign, manifests as easy bruising, bleeding, and petechiae. Underproduction of normal neutrophils predisposes patients to infections, such as pneumonias, tooth infections, and sinusitis.
Leukemia cell expansion within the marrow may lead to bone pain and, in young children, resistance to walking. Extramedullary deposition leads to lymphadenopathy, hepatosplenomegaly with abdominal tenderness to palpation, and testicular enlargement, while involvement of the CNS leads to headaches, nausea, vomiting, and cranial nerve palsies. A mediastinal mass, which may be seen in T-cell ALL, may result in chest discomfort, shortness of breath, dyspnea on exertion, and superior vena cava syndrome.
Rapidly proliferating disease may result in spontaneous tumor lysis with renal failure and electrolyte imbalances. Many patients present with fevers without infectious etiologies.
A summary of clinical features is presented in Table 28-2.
 DIAGNOSTIC STUDIES
DIAGNOSTIC STUDIES
The WHO classification describes three distinct entities of ALL: B-ALL with recurrent genetic abnormalities, B-ALL not otherwise specified (cytogenetically normal), and T-ALL. To classify a hematologic malignancy as ALL, there must be at least 25% involvement of precursor lymphoblasts committed to the B-cell or T-cell lineage present in the bone marrow and blood. This diagnostic criterion contrasts with AML, which requires ≥20% myeloblasts in the bone marrow or blood for diagnosis. Burkitt’s lymphoma is classified as a “mature” B-ALL and is the exception to this rule. Burkitt’s cells are negative for both myeloperoxidase (MPO) and TdT but stain positively for B-cell markers and, often, have light chain restriction.
The diagnostic workup consists of blood tests, imaging, bone marrow aspiration and biopsy, and lumbar puncture (LP) (Table 28-3). The CBC and peripheral blood smear may show leukocytosis with lymphoblasts (Figure 28-1) with decreases in normal blood counts. Serum chemistries reflect the degree of tumor burden and cell lysis; patients with tumor lysis exhibit hyperuricemia, hypocalcemia, hyperphosphatemia, hyperkalemia, and elevated LDH. Bone marrow aspiration reveals hypercellularity with increased lymphoblasts. CNS involvement is present in 5%–15% of adults and children alike, and is more frequently associated with the precursor T-cell immunophenotype. LP and subsequent CNS analysis will show blasts by cell count and cytology and may demonstrate an elevated opening pressure and protein and glucose derangements. Evidence exists that a traumatic LP may seed the CNS in unaffected children. Consequently, a traumatic tap as suggested by the presence of red blood cells in the cell analysis represents an indication for intensification of CNS therapy (5). An anterior mediastinal mass may be detected in 5%–10% of children and 15% of adults by chest X-ray, a finding more commonly associated with T-ALL.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


