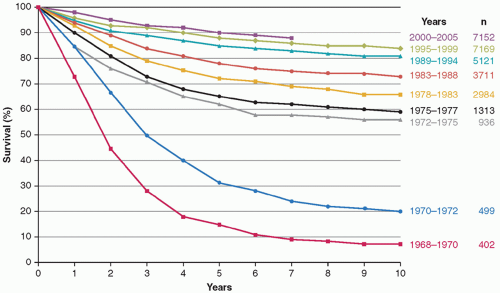
 Figure 19.1 Improvement in survival of children with ALL. Overall survival probability by treatment era for 29,287 children with ALL who enrolled on trials from 1968 to 2005 conducted by the Children’s Oncology Group (COG), Pediatric Oncology Group (POG), and Children’s Cancer Group (CCG). The 2000 to 2005 curve includes CCG, POG, and COG. The 1995 to 1999 curve includes CCG and POG. All other curves are CCG. (Adapted from Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol 2012;30:1663-1669.) |
with ALL each year in the United States, with an incidence of 2 to 5 cases per 100,000 U.S. children.7 The peak incidence of ALL occurs between 2 and 5 years of age. This is in marked contrast to acute myeloid leukemia (AML), which does not have a distinct peak age incidence in childhood (Fig. 19.2).
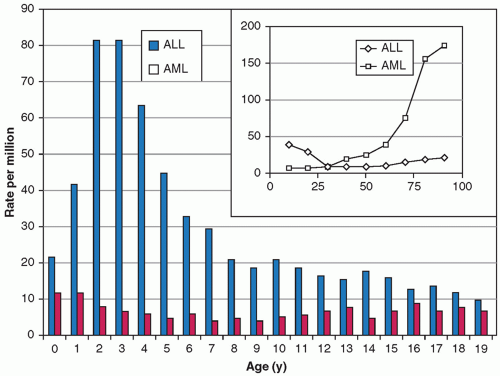 Figure 19.2 Age-specific incidence of ALL and AML in children and in adults (inset). (From Brown P, Hunger SP. Acute leukemia in children. In: Bope ET, Rakel RE, Kellerman RD, eds. Conn’s current therapy. Philadelphia, PA: Saunders, 2013:765-768, with permission.) |
 Figure 19.3 Kaplan-Meier estimates of EFS for a cohort of 8,447 children with ALL according to racial and ethnic distribution. (Adapted from Bhatia S, Sather HN, Heerema NA, et al. Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood 2002;100:1957-1964, with permission.) |
 Figure 19.4 Genetic ancestry and risk of relapse in childhood ALL. Higher levels of Native American (NA) ancestry were linked to increased risk of relapse in patients with native American ancestry who did not receive delayed intensification (A) but not in those who did receive delayed intensification in the Children’s Oncology Group P9904/9905 trial (B). Red = NA ancestry <10%, blue = NA ancestry >10%. (Adapted from Yang JJ, Cheng C, Devidas M. Ancestry and pharmacogenomics of relapse in acute lymphoblastic leukemia. Nat Genet 2011;43:237-241, with permission.) |
this type of ALL and neurologic abnormalities.30 Although AML is more common in Bloom syndrome as well as in patients with Fanconi anemia, ALL may occur.
TABLE 19.1 Selected Recurrent Genetic Alterations in Childhood Acute Lymphoblastic Leukemia (ALL) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
fields (EMFs), routine emissions from nuclear power plants, or fallout from atmospheric nuclear testing may be causally related to the development of childhood ALL have not withstood close scientific scrutiny.
combinations. The WHO classification recognizes two specific genetic subgroups, characterized by BCR-ABL or MLL rearrangement, and a third subgroup for cases with any other chromosomal abnormalities, subclassified by the lineages present. There is little systematic data about this rare form of leukemia, but outcomes are generally poor (see also Treatment section).61
TABLE 19.2 Monoclonal Antibodies Commonly Used to Immunophenotype Leukemia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
near-tetraploid, a distinct category in terms of chromosomal doubling pattern.66 Near-tetraploidy is often associated with T-ALL or ETV6-RUNX2+ B-ALL. Early studies suggested a poor outcome, but more recent data suggest a favorable prognosis for near-tetraploid ALL.67 Cases in the pseudodiploid category (those with a DI = 1.0 or normal chromosome number but other abnormalities) have a relatively poorer prognosis. Those with diploidy and low hyperdiploidy (48 to 50 chromosomes) have a slightly worse prognosis than high hyperdiploid cases. In childhood ALL, approximately 20% of cases are high hyperdiploid and 1% are hypodiploid (Fig. 19.5). In T-ALL, the majority of cases are pseudodiploid or diploid.
 Figure 19.5 Cytogenetic and molecular genetic abnormalities in childhood ALL. Acute lymphoblastic leukemia with rearrangement of CRLF2 but without the BCR-ABL1-like transcriptional profile rarely presents with other classifying karyotypic alterations, but can be noted with high hyperdiploidy. Dicentric cases might have a range of translocations, including classifying translocations (e.g., ETV6-RUNX1). iAMP21, intrachromosomal amplification of chromosome 21; ETP, early T-cell precursor. (From Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013;381:1943-1955, with permission.) |
stratified by modal chromosome number into near haploid (24 to 31), low hypodiploid (32 to 39), and high hypodiploid (40 to 43) (Fig. 19.8).31 Cases with any chromosome number <44 have a dismal EFS of approximately 30%, while cases with 44 chromosomes have a somewhat better EFS of approximately 50% (Fig. 19.9).71 Hypodiploidy with 45 chromosomes (frequently associated with dicentric chromosomes 9p and/or 12p) has a relatively good prognosis.72 TP53 mutations have been identified in 91% of low-hypodiploid ALL cases, with nearly half of these mutations occurring in the germline as well (Fig. 19.8C, D).31 Thus, identification of low hypodiploid ALL should prompt investigation of the patient’s tumor and germline TP53 status and family history for cancers characteristic of Li-Fraumeni syndrome. Special mention should be made of cases that undergo doubling of the hypodiploid clone. In some cases, both clones are present, and in others, referred to as “masked hypodiploidy,” only the doubled clone is retained. Since masked hypodiploidy does not appear to differ from nonmasked hypodiploidy in clinical or prognostic
features, it is crucial to distinguish it from hyperdiploidy, which in contrast bears a favorable prognosis and might prompt de-escalation of therapy.31,73 Suspicion may be raised by a predominance of tetrasomies affecting characteristic chromosomes (most often 8, 10, 14, 18, and 21). The diagnosis may be made by microsatellite panel or SNP array, which demonstrate LOH for the disomic chromosomes of the doubled clone.
 Figure 19.6 Application of new techniques for characterizing ALL. A: DNA copy number profiling in 242 pediatric ALL cases. Each case is represented by a column. Pink represents diploid copy number, white deletion, and red amplification. HD > 50, hyperdiploidy with greater than 50 chromosomes; Ph, BCR-ABL1 positive ALL; Hypo, B-precursor ALL with hypodiploidy. (From Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature 2007;446:758-764, with permission.) B: Whole-genome sequencing (WGS) identifies recurring mutations in T-ALL. Data are shown for 106 T-ALL cases, including 12 cases subjected to WGS (arrowed), and 94 recurrence cases. Cases are grouped by early T-precursor status. Genes identified as novel targets of mutation in T-ALL are labeled in green. (From Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukemia. Nature 2012;481:157-163, with permission.) C: Gene-expression profiling identifies a novel BCR-ABL1-like subtype of ALL (left column of gray bar indicates BCR-ABL1+ cases, and right column of gray bar indicates BCR-ABL1-like cases). (From Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009;10:125-134, with permission.) D: Genome-wide association study identifies germline single-nucleotide polymorphisms (SNPs) associated with susceptibility to childhood ALL. The x-axis shows the P values (-log10) for 307,944 germline SNPs. The allele frequency of the SNPs was compared between children with ALL in the discovery cohort (n = 317) and the combined non-ALL control cohort (n = 17,958). The SNP whose allele frequency differed most significantly between the two cohorts (p = 1.4 × 10-15) was localized to chromosome 10. The dashed line shows the threshold P value indicating genome-wide significance. (From Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 2009;41:1001-1005, with permission.) E: Mouse xenografts established by tail vein injection of luciferase-labeled leukemic blasts can be used to test in vivo efficacy of novel therapies. (Courtesy of Alexandra Stevens and Michele Redell, Baylor College of Medicine.) F: Phospho-flow cytometry can be used to demonstrate increased activity of specific signaling pathways in ALL blasts and to identify therapeutically useful agents that inhibit these pathways. |
 Figure 19.7 Kaplan-Meier analysis of event-free survival according to biological subtype of leukemia. (From Pui CH, Robison LL, Look AT. Acute lymphoblastic leukemia. Lancet 2008;371:1030-1043, with permission.) |
 Figure 19.8 Molecular genetics of hypodiploid ALL. A: Genome-wide DNA copy number heatmap showing SNP 6.0 microarray data for pediatric hypodiploid ALL cases. Chromosomes 1-22, X and Y are depicted from top to bottom, and individual samples are shown from left to right. LH, low hypodiploid. Blue indicates deletions and red indicates amplifications. B: Unsupervised principal components analysis (PCA) of gene-expression data from all hypodiploid ALL cases with available high-quality RNA (n = 94) distinguishes near-haploid, low-hypodiploid, and near-diploid ALL subgroups. Right, hierarchical clustering analysis of gene-expression array data. C: Frequent mutations in TP53 in pediatric hypodiploid ALL are indicated on the p53 protein domain plot. Known Li-Fraumeni syndrome (LFS) alterations are indicated in red. Alterations present in nontumor cells are indicated by blue lines. D: Pedigree of a family with an inherited TP53 mutation. N, number of siblings. (From Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013;43:242-252, with permission.) |
 Figure 19.9 Overall survival in hypodiploid ALL. Overall survival for 130 evaluable, non-Ph+ patients by modal chromosome number: 44 chromosomes, 40-43 chromosomes, 30-39 chromosomes, and 24-29 chromosomes. (Adapted from Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 2007;110:1112-1115, with permission.) |
 Figure 19.10 Outcomes in infant ALL. Event-free survival for infants treated on the Interfant-99 trial, by MLL status, age, and initial white blood count. Group 1: patients with germline MLL gene. Group 2: patients with MLL gene rearrangement, age <6 months at diagnosis and initial white blood count >300 109 cells/L. Group 3: all other patients. (From Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 2007;370:240-250, with permission.) |
forming a 185-to-190-kD protein usually known as p190. An additional breakpoint generates a 230-kD protein associated with a rare CML variant with neutrophilia and occasionally with classic CML. All three transcripts can be detected at very low levels using sensitive PCR techniques. It has been suggested that the p190 protein arises de novo, whereas the p210 protein may represent the blast crisis of a previously unrecognized CML. Other features that may distinguish cases that originated as CML include basophilia, marked splenomegaly, and persistence of the BCR-ABL1 fusion protein in hematopoietic precursor cells of all lineages following remission.
 Figure 19.11 Outcomes in Philadelphia chromosome acute lymphoblastic leukemia (Ph+ ALL). Comparison of disease-free survival in Ph+ ALL patients treated on Children’s Oncology Group study AALL0031 Cohort 5. Cohort 5 patients received continuous dosing of imatinib during intensive chemotherapy and intermittent dosing during maintenance therapy. The curves compare Cohort 5 patients who received chemotherapy only versus related and unrelated bone marrow transplant (BMT). (From Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: children’s Oncology Group study AALL0031. Leukemia 2014;28(7):1467-1471, with permission.) |
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


